scRNA-单细胞转录组数据校正批次效应实战
刘小泽写于19.6.23 文章来源:https://bioconductor.riken.jp/packages/3.7/workflows/vignettes/simpleSingleCell/inst/doc/work-5-mnn.html
什么是批次效应?
大型的单细胞测序项目一般都会产生许多细胞,这些样本制备过程很难保持时间一致、试剂一致,另外上机测序的时候也不一定在同一个测序仪上
具体可以看这篇文章:https://www.nature.com/articles/nrg2825
Batch effects are sub-groups of measurements that have qualitatively different behaviour across conditions and are unrelated to the biological or scientific variables in a study. For example, batch effects may occur if a subset of experiments was run on Monday and another set on Tuesday, if two technicians were responsible for different subsets of the experiments or if two different lots of reagents, chips or instruments were used.
简而言之,不同时间、不同操作者、不同试剂、不同仪器导致的实验误差,反映到细胞的表达量上就是批次效应,这个很难去除但可以缩小。如果效应比较还可以接受,但是批次效应很严重,就可能会和真实的生物学差异相混淆,让结果难以捉摸。我们需要辨别到底存在多大程度的批次效应,对我们真实的生物学样本会不会产生影响。
校正批次效应的目的就是:减少batch之间的差异,尽量让多个batch的数据重新组合在一起,这样下游分析就可以只考虑生物学差异因素
目前有种方法:removeBatchEffect()(Ritchie et al. 2015),它属于limma包,假定细胞群体组成在批次中是已知或相同的 https://dash.harvard.edu/bitstream/handle/1/15034877/4402510.pdf?sequence=1
本文中使用的方法是mnnCorreect() (Haghverdi et al. 2018) https://www.nature.com/articles/nbt.4091,它基于高维表达空间中最近邻居(MNN)检测的批量修正策略,不需要预先定义或者已知全部细胞群体组成,它只需要在批次之间有关联的一小部分群体
下面将会利用三个人类胰腺scRNA-seq数据集,而且它们来自不同的组,使用不同的方法得到,因此预测会存在较大的批次效应
处理不同的数据集
第一个数据:CEL-seq, GSE81076
加载数据
数据是Grun et al. (2016) 利用CEL-seq方法,加入了UMI、ERCC,表达矩阵可以从GEO获取(https://www.ncbi.nlm.nih.gov//geo/query/acc.cgi?acc=GSE81076)
这里要学会组织R数据的方式:先新建一个文件夹,随便从其他地方复制一个
.Rpoj到这个目录,修改一下名称(最好将文件夹和这个文件都设置为英文)。双击打开.Rpoj就好,那么以后直接将数据下载到这个目录下,它就成为了我们整个项目的"根据地“
# 首先清空变量和设置默认不要因子型
rm(list = ls())
options(stringsAsFactors = F)
# 压缩文件可以直接读取
gse81076.df <- read.table("GSE81076_D2_3_7_10_17.txt.gz", sep='\t',
header=TRUE, row.names=1)
> dim(gse81076.df)
[1] 20148 1728
看一下样本信息
很不幸,这个数据的列名是压缩的,包含了多种注释信息
> head(colnames(gse81076.df))
[1] "D2ex_1" "D2ex_2" "D2ex_3" "D2ex_4" "D2ex_5" "D2ex_6"
我是怎么发现的? 因为GEO描述中写道donor的编号就这么几种,因此列名的
D加紧随的数字就是donor信息

工欲善其事必先利其器
需要使用sub,不理解可以看一下:https://en.wikibooks.org/wiki/R_Programming/Text_Processing#Detecting_the_presence_of_a_substring
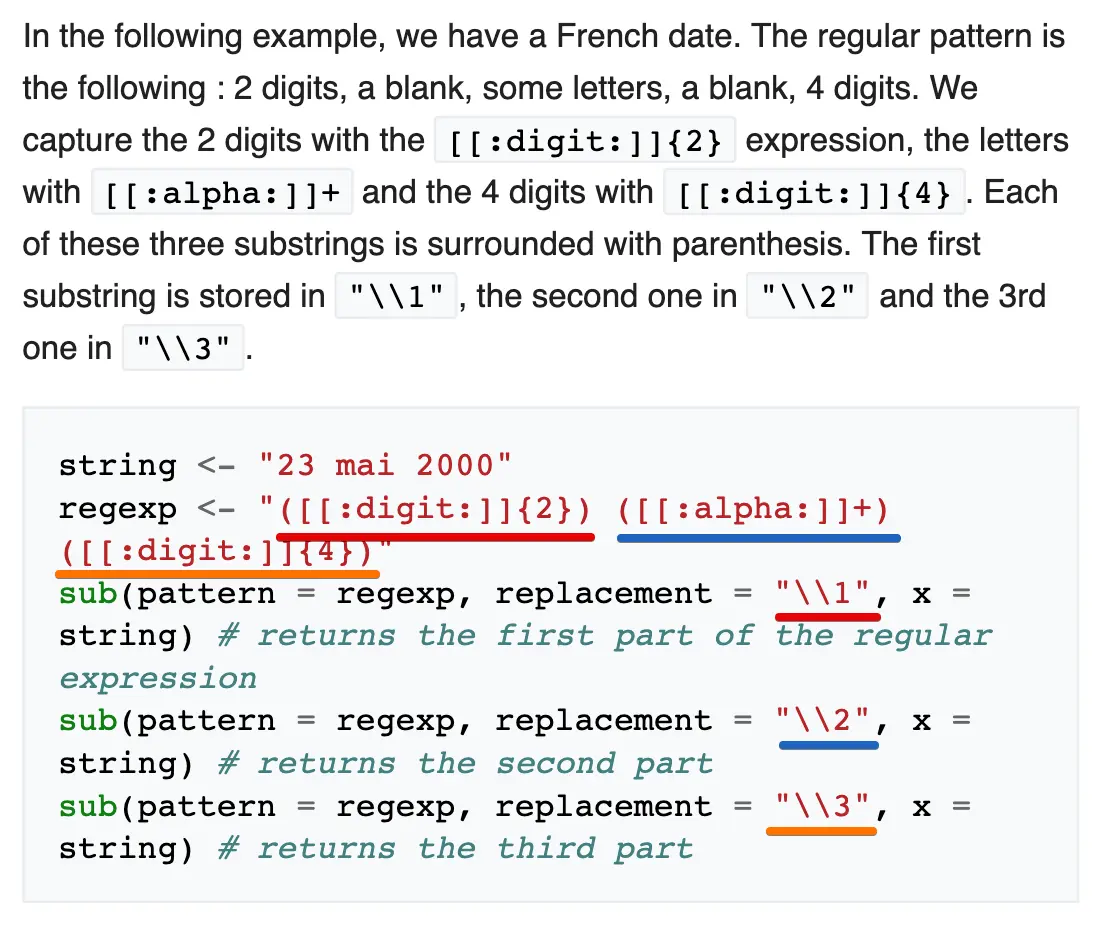
sub包含三部分:要匹配的内容、要替换成的内容、操作的内容,两种形式:
# 第一种:不利用正则
> text <- "abc def ghk"
> sub(pattern = " ", replacement = "", x = text)
[1] "abcdef ghk" #可以看到,要匹配空格,替换为无(也就是去掉空格)
# 第二种:利用正则匹配
sub(pattern = regexp, replacement = "\\1", x = string)
# 结果返回就是regexp中第一个小括号包围的内容,同理\\2返回第二个小括号返回的内容
最后注意一下:
sub是替换第一个匹配到的内容,gsub是一次性全部替换匹配到的内容(即:global substitute)
好,了解了需要用的工具,先将donor ID提取出来:
donor.names <- sub("^(D[0-9]+).*", "\\1", colnames(gse81076.df))
# 理解一下这个代码:
# 先看第一部分,^(D[0-9]+).* 这是利用正则表达式匹配的,主要看小括号,这里的是一会要返回的内容。意思就是说D后面是0-9数字,可以是1个或多个
# 检查一下
> table(donor.names)
donor.names
D101 D102 D10631 D17 D1713 D172444 D2 D3 D71
96 96 96 288 96 96 96 480 96
D72 D73 D74
96 96 96
同理,列名中还包括了细胞板ID,也利用sub提取
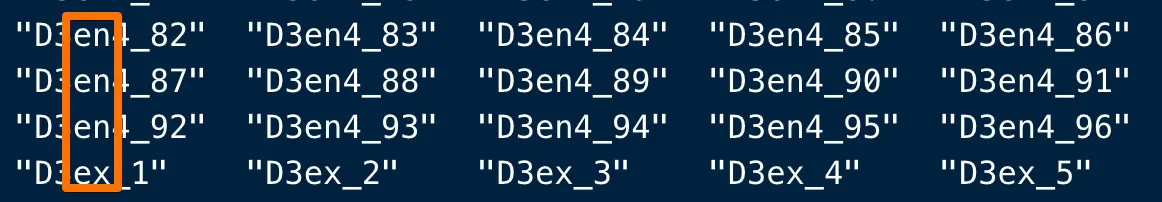
# 可以看到这个ID位于donor ID后面(当然有的样本名不包含plate ID)
plate.id <- sub("^D[0-9]+(.*)_.*", "\\1", colnames(gse81076.df))
# 还是看小括号中包含的,意思是位于donor ID和下划线之前,可以是0到多个字符
# 检查一下
> table(plate.id)
plate.id
All1 All2 en1 en2 en3 en4 ex TGFB
864 96 96 96 96 96 96 192 96
看一下基因信息
同样是比较混乱的,并不是我们想要的标准名称(如Ensembl ID、gene symbol等)
> head(rownames(gse81076.df))
[1] "A1BG-AS1__chr19" "A1BG__chr19" "A1CF__chr10" "A2M-AS1__chr12"
[5] "A2ML1__chr12" "A2MP1__chr12"
看起来这个比较容易替换,只要将__chr及后面的内容去掉就好(采用上面所写的sub第一种方法即可),并且注意是gsub ,全部替换的意思
gene.symb <- gsub("__chr.*$", "", rownames(gse81076.df))
# 注意要加一个结尾标志符号 $
看看有没有ERCC
目前已经有了基因名,那么看看有没有ERCC,直接grep搜索即可
需要注意的是,grep返回的结果是一个字符串(也就是匹配到的ERCC位置);另外grepl返回逻辑值,也就是全部基因中哪些是ERCC(标记TRUE),哪些不是(标记为FALSE)
is.spike <- grepl("^ERCC-", gene.symb) #结果返回大量的TRUE和FALSE
> table(is.spike)
is.spike
FALSE TRUE
20064 84
# 看到共有84个ERCC
# 以前10列为例,看看ERCC内容
View(gse81076.df[grep("^ERCC-", gene.symb),1:10])
高ERCC含量与低质量数据相关,通常是排除的标准。https://scrnaseq-course.cog.sanger.ac.uk/website/cleaning-the-expression-matrix.html也有提及:

如果ERCC的reads数很高,则表示起始内源性RNA总量低(可能发生了细胞凋亡或者其他胁迫因素导致的RNA降解;另外还可能是细胞体积小,一般来讲小细胞比大细胞有更高比例的ERCC)。
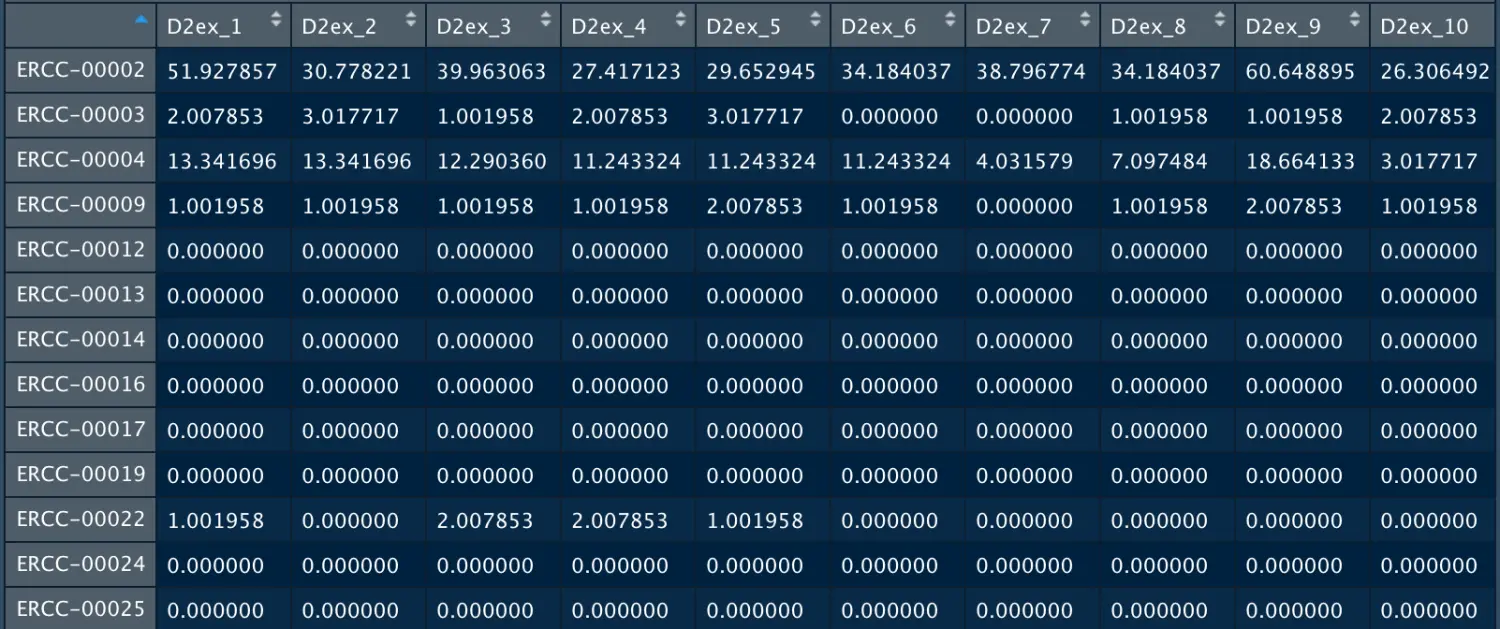
可能你会想,ERCC是内参基因吗? 其实并不是,它相对于内参基因会更稳定,看:https://cofactorgenomics.com/6-changes-thatll-make-big-difference-rna-seq-part-3/ Spike-in controls are inherently advantageous to endogenous housekeeping genes for normalization, as potential housekeeping genes such as ACTB, GAPDH, HPRT1, and B2M, etc. vary considerably under different experimental conditions
转化基因ID
library(org.Hs.eg.db)
gene.ids <- mapIds(org.Hs.eg.db, keys=gene.symb, keytype="SYMBOL", column="ENSEMBL")
> length(gene.ids)
[1] 20148
> length(gene.symb)
[1] 20148
> head(gene.symb)
[1] "A1BG-AS1" "A1BG" "A1CF" "A2M-AS1" "A2ML1" "A2MP1"
> head(gene.ids)
A1BG-AS1 A1BG A1CF A2M-AS1
"ENSG00000268895" "ENSG00000121410" "ENSG00000148584" "ENSG00000245105"
A2ML1 A2MP1
"ENSG00000166535" "ENSG00000256069"
转换完id后,记住:这里代码只是将标准的基因名转换过去,还有一些ERCC没有转换,找个例子就知道,在新的gene.ids中ERCC全部显示为NA
> grep("^ERCC-",gene.symb)
[1] 5179 5180 5181 5182 5183 5184 5185 5186 5187 5188 5189 5190 5191 5192
[15] 5193 5194 5195 5196 5197 5198 5199 5200 5201 5202 5203 5204 5205 5206
[29] 5207 5208 5209 5210 5211 5212 5213 5214 5215 5216 5217 5218 5219 5220
[43] 5221 5222 5223 5224 5225 5226 5227 5228 5229 5230 5231 5232 5233 5234
[57] 5235 5236 5237 5238 5239 5240 5241 5242 5243 5244 5245 5246 5247 5248
[71] 5249 5250 5251 5252 5253 5254 5255 5256 5257 5258 5259 5260 5261 5262
> gene.ids[5179]
ERCC-00002
NA
# 使用一句代码即可替换
gene.ids[is.spike] <- gene.symb[is.spike]
# 再检查一下
> gene.ids[5179]
ERCC-00002
"ERCC-00002"
去重复基因和没有表达量的(NA)
# 去重复和NA(这里采用反选的方法)
keep <- !is.na(gene.ids) & !duplicated(gene.ids)
gse81076.df <- gse81076.df[keep,]
rownames(gse81076.df) <- gene.ids[keep]
> summary(keep)
Mode FALSE TRUE
logical 2071 18077
# 结果过滤掉了2071个基因
创建单细胞对象
创建SingleCellExperiment这个对象,将count矩阵和注释信息放在一起
# 创建对象存储count和metadata
library(SingleCellExperiment)
sce.gse81076 <- SingleCellExperiment(list(counts=as.matrix(gse81076.df)),
colData=DataFrame(Donor=donor.names, Plate=plate.id),
rowData=DataFrame(Symbol=gene.symb[keep]))
# 重新设定一下ERCC位置
isSpike(sce.gse81076, "ERCC") <- grepl("^ERCC-", rownames(gse81076.df))
# 看下结果
> sce.gse81076
class: SingleCellExperiment
dim: 18077 1728
metadata(0):
assays(1): counts
rownames(18077): ENSG00000268895 ENSG00000121410 ... ENSG00000074755
ENSG00000036549
rowData names(1): Symbol
colnames(1728): D2ex_1 D2ex_2 ... D17TGFB_95 D17TGFB_96
colData names(2): Donor Plate
reducedDimNames(0):
spikeNames(1): ERCC
质控和标准化
对每个细胞进行质控,并鉴定出文库很小/表达基因少/ERCC含量高的细胞
library(scater)
sce.gse81076 <- calculateQCMetrics(sce.gse81076, compact=TRUE)
QC <- sce.gse81076$scater_qc
low.lib <- isOutlier(QC$all$log10_total_counts, type="lower", nmad=3) # 这个nmad意思就是偏离MAD计算结果几位,被认为是偏离值(用lower向左找更小的,higher向右找更大的)
low.genes <- isOutlier(QC$all$log10_total_features_by_counts, type="lower", nmad=3)
high.spike <- isOutlier(QC$feature_control_ERCC$pct_counts, type="higher", nmad=3)
data.frame(LowLib=sum(low.lib), LowNgenes=sum(low.genes),
HighSpike=sum(high.spike, na.rm=TRUE))
# 看下结果
LowLib LowNgenes HighSpike
1 55 130 388
绝对中位差(MADs)是用原数据减去中位数后得到的新数据的绝对值的中位数,可以用来估计标准差
上面返回的结果,被认为是低质量的样本数据,需要被移除
另外还有许多QC的检验标准,后续再探索
discard <- low.lib | low.genes | high.spike
sce.gse81076 <- sce.gse81076[,!discard]
> summary(discard)
Mode FALSE TRUE
logical 1292 436
# 看到总共过滤掉了400多个细胞
正式标准化之前需要先做几项工作:
先进行细胞聚类,利用
quickCluster(),这样做可以避免将差异很大的细胞混在一起导致误差。通过看帮助文档得知,这个函数有两种方法:hclust和igraph,基于spearman相关性分析,这种方法相对于pearson方法,是非线性的,可以针对不同量纲数据计算。我们只关心每个数值在变量内的排列顺序,如果两个变量的对应值,在各组内的排位顺序是相同或类似的,那么就认为有显著地相关性library(scran) clusters <- quickCluster(sce.gse81076, min.mean=0.1) > table(clusters) clusters 1 2 3 4 445 356 252 239专用的标准化方法:之前听过的CPM/FPKM/TPM/TMM都是适用于bulk RNA-seq,分析时也经常移植到单细胞数据。但是单细胞数据有自己的特点,例如存在细胞差异和基因差异两类系统偏差,而上述方法主要考虑了基因差异。为了校正细胞间的差异,
scran包特意利用去卷积法(deconvolution) 开发了computeSumFactors函数(Lun, Bach, and Marioni 2016)。它将聚类后的多组细胞合并在一起屏蔽0值分散的问题,并采用类似CPM的方法计算标准化因子(size factor)关于scran的描述:https://www.stephaniehicks.com/2018-bioinfosummer-scrnaseq/cleaning-the-expression-matrix.html
scranpackage implements a variant on CPM specialized for single-cell data (L. Lun, Bach, and Marioni 2016). Briefly this method deals with the problem of vary large numbers of zero values per cell by pooling cells together calculating a normalization factor (similar to CPM) for the sum of each pool. Since each cell is found in many different pools, cell-specific factors can be deconvoluted from the collection of pool-specific factors using linear algebra.补充:CPM为原始reads除以一个样品总的可用reads数乘以
1,000,000,但这种方法容易受到极高表达且在不同样品中存在差异表达的基因的影响,有点"牵一发动全身"的感觉代码实现
sce.gse81076 <- computeSumFactors(sce.gse81076, min.mean=0.1, clusters=clusters) # 另外对ERCC也单独计算size factor(用general.use=FALSE表示单独计算) sce.gse81076 <- computeSpikeFactors(sce.gse81076, general.use=FALSE) # 最后计算标准化的log表达量,用作下游分析 sce.gse81076 <- normalize(sce.gse81076)
鉴定高变异基因
利用函数trendVar()和decomposeVar() 根据表达量计算highly variable genes (HVGs) ,并利用spike-in的偏差为技术噪声提供参考(因为本来应该很稳定的spike-in如果方差很大,也就是波动很明显,说明实验或测序环节出了问题)
设置block是为了确保我们不感兴趣的差异(比如plate、donor之间)不会扩大真实数据偏差
block <- paste0(sce.gse81076$Plate, "_", sce.gse81076$Donor)
两行代码进行统计
fit <- trendVar(sce.gse81076, block=block, parametric=TRUE)
dec <- decomposeVar(sce.gse81076, fit)
最后作图表示
plot(dec$mean, dec$total, xlab="Mean log-expression",
ylab="Variance of log-expression", pch=16)
OBis.spike <- isSpike(sce.gse81076)
points(dec$mean[is.spike], dec$total[is.spike], col="red", pch=16)
curve(fit$trend(x), col="dodgerblue", add=TRUE)
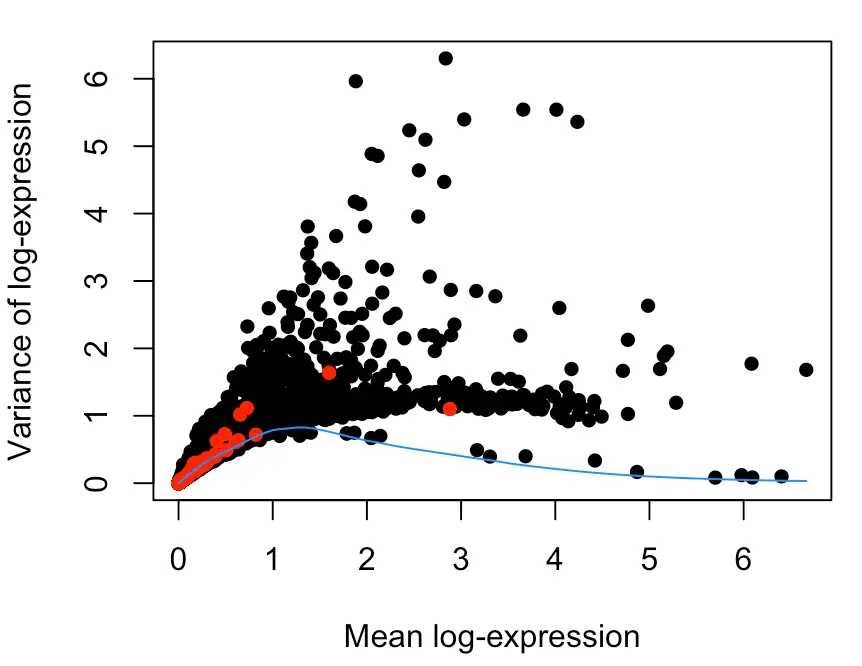
这张图不简单,我理解到的信息如下:
- 每个点表示一个基因;横坐标表示基因表达量高低(因为用log进行了缩放,所以标度变小);纵坐标表示基因表达量的波动大小;红点表示spike-in;蓝色线表示针对spike-in的波动情况做的趋势线
- 首先大部分红点在横坐标的1附近,表达量普遍处于低水平,按照之前介绍的:高ERCC含量与低质量数据相关,那么ERCC一般保持低水平就表示数据不错
- 然后大部分红点在纵坐标的1附近,表示方差(波动情况)并不大,因此可以认为技术误差在可控范围内(如果说存在很大的技术误差,那么应该会有几个红点在纵坐标较大的位置,然后趋势线也会比较陡峭)
- 在横坐标2-4这个范围内,一部分黑点(也就是一些基因)的纵坐标很大,表示它们在样本中的离散程度很大,有的样本中该基因表达量很小,有的却相当大,这就是部分要找的HVGs
挑出HVGs
在decomposeVar函数帮助文档中有一句描述:Highly variable genes (HVGs) can be identified as those with large biological components. The biological component is determined by subtracting the technical component from the total variance. (HVGs是具有高的biological components数值,而biological components取决于从总体方差中减去技术因素导致的方差)
dec.gse81076 <- dec
dec.gse81076$Symbol <- rowData(sce.gse81076)$Symbol
dec.gse81076 <- dec.gse81076[order(dec.gse81076$bio, decreasing=TRUE),]
> head(dec.gse81076,2)
DataFrame with 2 rows and 7 columns
mean total bio
<numeric> <numeric> <numeric>
ENSG00000254647 2.83712754306791 6.30184692631371 5.85904290864641
ENSG00000129965 1.88188510741958 5.96360144483475 5.5152391307155
tech p.value FDR Symbol
<numeric> <numeric> <numeric> <character>
ENSG00000254647 0.442804017667299 0 0 INS
ENSG00000129965 0.448362314119254 0 0 INS-IGF2
# 可以看到这里表达量变化最大的基因是INS,和胰岛素相关
刘小泽写于19.6.24
第二个数据–CEL-seq2, GSE85241
Muraro et al. (2016) 利用CEL-seq2技术并结合UMI、ERCC得到的
https://www.ncbi.nlm.nih.gov//geo/query/acc.cgi?acc=GSE85241
下面快速使用代码
读数据,看数据
gse85241.df <- read.table("GSE85241_cellsystems_dataset_4donors_updated.csv.gz", sep='\t', header=TRUE, row.names=1)
> dim(gse85241.df)
[1] 19140 3072
提取meta信息
# 还是先看一下
> head(colnames(gse85241.df))
[1] "D28.1_1" "D28.1_2" "D28.1_3" "D28.1_4" "D28.1_5" "D28.1_6"
# 依然是:点号前面的是donor信息
donor.names <- sub("^(D[0-9]+).*", "\\1", colnames(gse85241.df))
> table(donor.names)
donor.names
D28 D29 D30 D31
768 768 768 768
# 然后文章使用了8个96孔板,于是可以将点号和下划线之间的数字提取出来
plate.id <- sub("^D[0-9]+\\.([0-9]+)_.*", "\\1", colnames(gse85241.df)) #这句代码中注意使用了一个转义符\\,在R中需要用两个反斜线来表示转义
> table(plate.id)
plate.id
1 2 3 4 5 6 7 8
384 384 384 384 384 384 384 384
提取基因、ERCC信息
gene.symb <- gsub("__chr.*$", "", rownames(gse85241.df))
is.spike <- grepl("^ERCC-", gene.symb)
> table(is.spike)
is.spike
FALSE TRUE
19059 81
基因转换
library(org.Hs.eg.db)
gene.ids <- mapIds(org.Hs.eg.db, keys=gene.symb, keytype="SYMBOL", column="ENSEMBL")
gene.ids[is.spike] <- gene.symb[is.spike]
keep <- !is.na(gene.ids) & !duplicated(gene.ids)
gse85241.df <- gse85241.df[keep,]
rownames(gse85241.df) <- gene.ids[keep]
> summary(keep)
Mode FALSE TRUE
logical 1949 17191
# 去掉了快2000个重复或无表达的基因
创建单细胞对象
# 存储metadata作为colData、基因信息作为rawData、ERCC作为spike-in
sce.gse85241 <- SingleCellExperiment(list(counts=as.matrix(gse85241.df)),
colData=DataFrame(Donor=donor.names, Plate=plate.id),
rowData=DataFrame(Symbol=gene.symb[keep]))
isSpike(sce.gse85241, "ERCC") <- grepl("^ERCC-", rownames(gse85241.df))
质控和标准化
sce.gse85241 <- calculateQCMetrics(sce.gse85241, compact=TRUE)
QC <- sce.gse85241$scater_qc
low.lib <- isOutlier(QC$all$log10_total_counts, type="lower", nmad=3)
low.genes <- isOutlier(QC$all$log10_total_features_by_counts, type="lower", nmad=3)
high.spike <- isOutlier(QC$feature_control_ERCC$pct_counts, type="higher", nmad=3)
data.frame(LowLib=sum(low.lib), LowNgenes=sum(low.genes),
HighSpike=sum(high.spike, na.rm=TRUE))
# LowLib LowNgenes HighSpike
# 577 669 696
# 然后去掉低质量的细胞
discard <- low.lib | low.genes | high.spike
sce.gse85241 <- sce.gse85241[,!discard]
> summary(discard)
Mode FALSE TRUE
logical 2346 726
可以看到文库小的有577个,基因表达少的有669个,高spike-in的有696个,但是最后只去掉了726个,这是因为,有的细胞同时存在以上两种或三种低质量情况,因此并不能简单认为总共去除细胞数=577+669+696
聚类
clusters <- quickCluster(sce.gse85241, min.mean=0.1, method="igraph")
> table(clusters)
clusters
1 2 3 4 5 6
237 248 285 483 613 480
标准化
sce.gse85241 <- computeSumFactors(sce.gse85241, min.mean=0.1, clusters=clusters)
summary(sizeFactors(sce.gse85241))
sce.gse85241 <- computeSpikeFactors(sce.gse85241, general.use=FALSE)
summary(sizeFactors(sce.gse85241, "ERCC"))
sce.gse85241 <- normalize(sce.gse85241)
鉴定HVGs
block <- paste0(sce.gse85241$Plate, "_", sce.gse85241$Donor)
fit <- trendVar(sce.gse85241, block=block, parametric=TRUE)
dec <- decomposeVar(sce.gse85241, fit)
plot(dec$mean, dec$total, xlab="Mean log-expression",
ylab="Variance of log-expression", pch=16)
is.spike <- isSpike(sce.gse85241)
points(dec$mean[is.spike], dec$total[is.spike], col="red", pch=16)
curve(fit$trend(x), col="dodgerblue", add=TRUE)
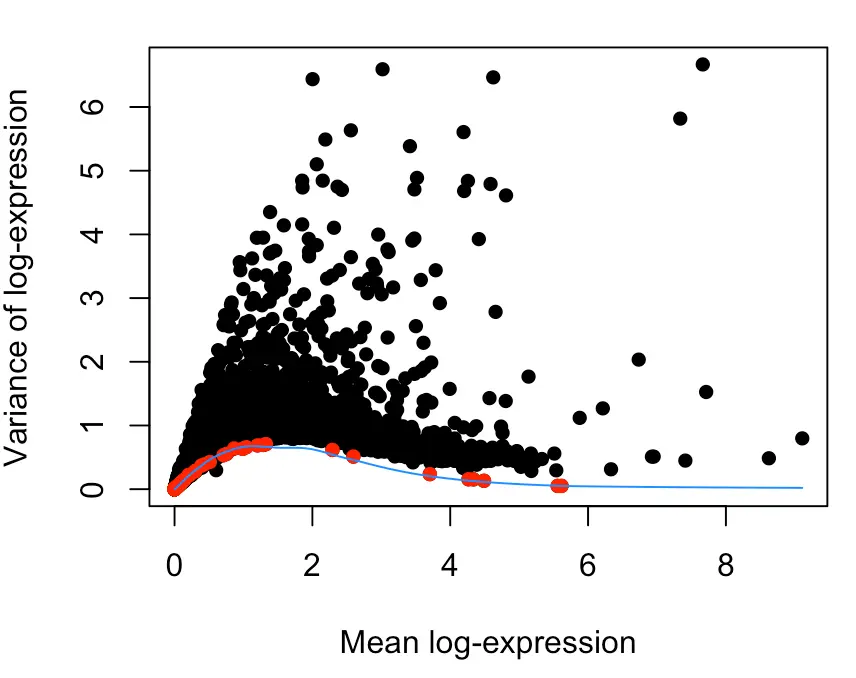
这张图中的ERCC表达量就有一些比较高的,但是占比不高,另外总体波动不大
# 选出来这些基因
dec.gse85241 <- dec
dec.gse85241$Symbol <- rowData(sce.gse85241)$Symbol
dec.gse85241 <- dec.gse85241[order(dec.gse85241$bio, decreasing=TRUE),]
> head(dec.gse85241,2)
DataFrame with 2 rows and 7 columns
mean total bio
<numeric> <numeric> <numeric>
ENSG00000115263 7.66453729345785 6.66863456231166 6.63983282676052
ENSG00000089199 4.62375793902937 6.46558866721711 6.34422879524839
tech p.value FDR Symbol
<numeric> <numeric> <numeric> <character>
ENSG00000115263 0.0288017355511366 0 0 GCG
ENSG00000089199 0.12135987196872 0 0 CHGB
第三个数据–Smart-seq2, E-MTAB-5061
Segerstolpe et al. (2016)利用Smart-seq2,添加了ERCC,这个数据和上面两个不同,它存放在ArrayExpress数据库,当然也是用链接规律的:https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5061/ (这个文件比较大,压缩文件151M,解压后700多M)

读入数据
文件较大,先读入样本,也就是第一行(nrow=1),看下数量
header <- read.table("pancreas_refseq_rpkms_counts_3514sc.txt",
nrow=1, sep="\t", comment.char="")
# 先看下header信息
> header[1,1:4]
V1 V2 V3 V4
#samples HP1502401_N13 HP1502401_D14 HP1502401_F14
# 然后将第一个(#samples)去掉
ncells <- ncol(header) - 1L #保存为整数
然后只加载基因名称和表达矩阵
# 这段代码需要再好好理解下
col.types <- vector("list", ncells*2 + 2)
col.types[1:2] <- "character"
col.types[2+ncells + seq_len(ncells)] <- "integer"
e5601.df <- read.table("pancreas_refseq_rpkms_counts_3514sc.txt",
sep="\t", colClasses=col.types)
# 最后将基因信息和表达矩阵分离
gene.data <- e5601.df[,1:2]
e5601.df <- e5601.df[,-(1:2)]
colnames(e5601.df) <- as.character(header[1,-1])
dim(e5601.df)
## [1] 26271 3514
判断ERCC
# gene.data[,2]对应测序数据中的基因ID,gene.data[,1]是相应的symbol ID
is.spike <- grepl("^ERCC-", gene.data[,2])
> table(is.spike)
is.spike
FALSE TRUE
26179 92
基因转换
library(org.Hs.eg.db)
gene.ids <- mapIds(org.Hs.eg.db, keys=gene.data[,1], keytype="SYMBOL", column="ENSEMBL")
gene.ids[is.spike] <- gene.data[is.spike,2]
# 去掉重复和无表达基因
keep <- !is.na(gene.ids) & !duplicated(gene.ids)
e5601.df <- e5601.df[keep,]
rownames(e5601.df) <- gene.ids[keep]
> summary(keep)
Mode FALSE TRUE
logical 3367 22904
提取metadata信息
metadata <- read.table("E-MTAB-5061.sdrf.txt", header=TRUE,
sep="\t", check.names=FALSE)
m <- match(colnames(e5601.df), metadata$`Assay Name`)
stopifnot(all(!is.na(m)))
metadata <- metadata[m,]
donor.id <- metadata[["Characteristics[individual]"]]
> table(donor.id)
donor.id
AZ HP1502401 HP1504101T2D HP1504901 HP1506401
96 352 383 383 383
HP1507101 HP1508501T2D HP1509101 HP1525301T2D HP1526901T2D
383 383 383 384 384
创建单细胞对象
sce.e5601 <- SingleCellExperiment(list(counts=as.matrix(e5601.df)),
colData=DataFrame(Donor=donor.id),
rowData=DataFrame(Symbol=gene.data[keep,1]))
isSpike(sce.e5601, "ERCC") <- grepl("^ERCC-", rownames(e5601.df))
后面的操作和之前保持一致了
sce.e5601 <- calculateQCMetrics(sce.e5601, compact=TRUE)
QC <- sce.e5601$scater_qc
low.lib <- isOutlier(QC$all$log10_total_counts, type="lower", nmad=3)
low.genes <- isOutlier(QC$all$log10_total_features_by_counts, type="lower", nmad=3)
high.spike <- isOutlier(QC$feature_control_ERCC$pct_counts, type="higher", nmad=3)
low.spike <- isOutlier(QC$feature_control_ERCC$log10_total_counts, type="lower", nmad=2)
data.frame(LowLib=sum(low.lib), LowNgenes=sum(low.genes),
HighSpike=sum(high.spike, na.rm=TRUE), LowSpike=sum(low.spike))
# LowLib LowNgenes HighSpike LowSpike
# 162 572 904 359
# 舍弃低质量细胞
discard <- low.lib | low.genes | high.spike | low.spike
sce.e5601 <- sce.e5601[,!discard]
> summary(discard)
Mode FALSE TRUE
logical 2285 1229
# 聚类
clusters <- quickCluster(sce.e5601, min.mean=1, method="igraph")
> table(clusters)
clusters
1 2 3 4 5 6
305 307 469 272 494 438
# 标准化
sce.e5601 <- computeSumFactors(sce.e5601, min.mean=1, clusters=clusters)
sce.e5601 <- computeSpikeFactors(sce.e5601, general.use=FALSE)
sce.e5601 <- normalize(sce.e5601)
因为这个数据中donor信息比较多,所以可视化也要特别对待
donors <- sort(unique(sce.e5601$Donor))
> donors
[1] "AZ" "HP1502401" "HP1504101T2D" "HP1504901"
[5] "HP1506401" "HP1507101" "HP1508501T2D" "HP1509101"
[9] "HP1525301T2D" "HP1526901T2D"
一共10个donor,作图可以设置这个参数,调整图片为2列
par(mfrow=c(ceiling(length(donors)/2), 2),
mar=c(4.1, 4.1, 2.1, 0.1))
代码作图,注意这段代码和之前的不同
collected <- list() # 第一行可以先不管,目的是创建一个空列表
# 下面进行一个循环,对10个donor进行循环:先取出第一个donor的列信息,然后使用if判断它是不是大于两列(也就是说:这个donor是不是有两个以上的细胞样本),如果只有一列那么就舍去;然后对这个donor的所有列进行标准化,去掉细胞文库差异;接着利用trendVar和decomposeVar鉴定HVGs,然后和之前一样进行可视化;最后将这个donor鉴定出来的HVGs信息放入collected这个列表中,留着以后用
for (x in unique(sce.e5601$Donor)) {
current <- sce.e5601[,sce.e5601$Donor==x]
if (ncol(current)<2L) { next }
current <- normalize(current)
fit <- trendVar(current, parametric=TRUE)
dec <- decomposeVar(current, fit)
plot(dec$mean, dec$total, xlab="Mean log-expression",
ylab="Variance of log-expression", pch=16, main=x)
points(fit$mean, fit$var, col="red", pch=16)
curve(fit$trend(x), col="dodgerblue", add=TRUE)
collected[[x]] <- dec
}
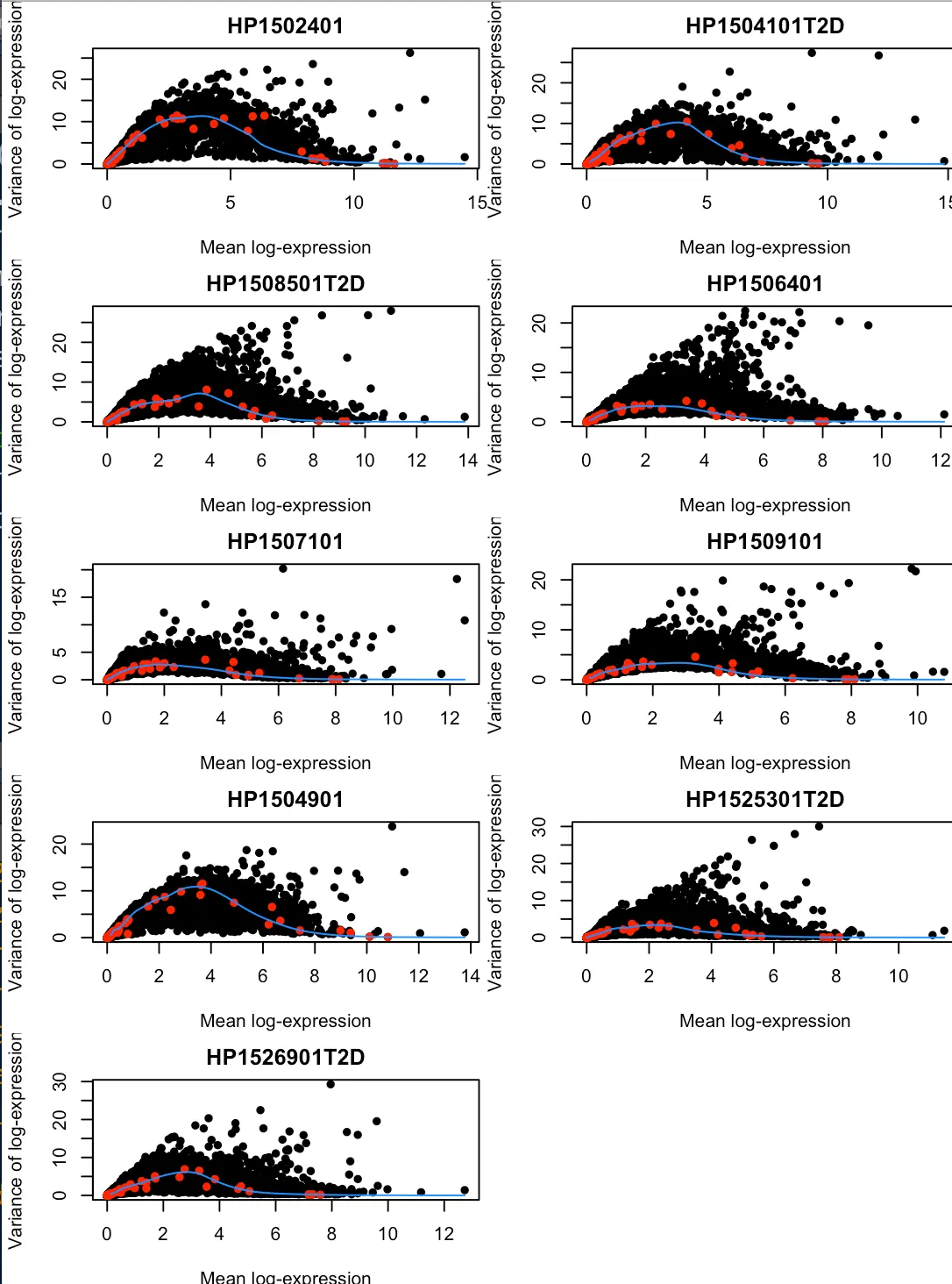
因为这个数据中donor信息比较多,因此我们需要将不同donor的HVGs整合成一个数据框(注意是更高级的SV4数据框)
dec.e5601 <- do.call(combineVar, collected)
dec.e5601$Symbol <- rowData(sce.e5601)$Symbol
dec.e5601 <- dec.e5601[order(dec.e5601$bio, decreasing=TRUE),]
> head(dec.e5601,3)
DataFrame with 3 rows and 7 columns
mean total bio
<numeric> <numeric> <numeric>
ENSG00000115263 9.79547495804957 24.9059740209558 24.693297105741
ENSG00000118271 10.3601718361198 19.0590510324402 18.9670050741979
ENSG00000089199 8.78499018265489 17.2605488560106 16.9971950283286
tech p.value FDR Symbol
<numeric> <numeric> <numeric> <character>
ENSG00000115263 0.212676915214769 0 0 GCG
ENSG00000118271 0.0920459582422512 0 0 TTR
ENSG00000089199 0.263353827682004 0 0 CHGB
刘小泽写于19.6.30 这次结合之前的3个数据集筛选出来的HVGs,看看放在一起怎么处理批次效应
三组不同数据的混合
我们可以从每个数据集(也就是每个批次)中挑选前1000个生物学差异最大的基因
还记得之前是如何得到每个数据集的HVGs吗?主要利用
trendVar、decomposeVar,另外存在多个样本使用combineVar进行合并
整合ID
整合三个数据集的前1000基因后,我们用Reduce()对它们取基因名的交集,最后给基因交集寻找搭配的gene symbol
rm(list = ls())
options(stringsAsFactors = F)
load("gse81076.Rdata")
load("gse85241.Rdata")
load("e5601.Rdata")
# 选择前1000基因
top.e5601 <- rownames(dec.e5601)[seq_len(1000)]
top.gse85241 <- rownames(dec.gse85241)[seq_len(1000)]
top.gse81076 <- rownames(dec.gse81076)[seq_len(1000)]
# https://www.r-bloggers.com/intersect-for-multiple-vectors-in-r/
chosen <- Reduce(intersect, list(top.e5601, top.gse85241, top.gse81076))
# 添加gene symbol
symb <- mapIds(org.Hs.eg.db, keys=chosen, keytype="ENSEMBL", column="SYMBOL")
> DataFrame(ID=chosen, Symbol=symb)
DataFrame with 353 rows and 2 columns
ID Symbol
<character> <character>
1 ENSG00000115263 GCG
2 ENSG00000118271 TTR
3 ENSG00000089199 CHGB
4 ENSG00000169903 TM4SF4
5 ENSG00000166922 SCG5
... ... ...
349 ENSG00000087086 FTL
350 ENSG00000149485 FADS1
351 ENSG00000162545 CAMK2N1
352 ENSG00000170348 TMED10
353 ENSG00000251562 MALAT1
另外,还有一种取交集的方法:先将全部的进行Reduce(),再组合选择前1000
in.all <- Reduce(intersect, list(rownames(dec.e5601),
rownames(dec.gse85241), rownames(dec.gse81076)))
# 设置权重weighted=FALSE ,认为每个batch的贡献一致
combined <- combineVar(dec.e5601[in.all,], dec.gse85241[in.all,],
dec.gse81076[in.all,], weighted=FALSE)
chosen2 <- rownames(combined)[head(order(combined$bio, decreasing=TRUE), 1000)]
取交集的方法会了,但是有个问题不知你有没有注意到:
取交集前提是三个批次之间有相同的HVGs,但是如果对于不同细胞类型的marker基因,它们特异性较强,不一定会出现在所有的batch中
只不过,我们这里只关注交集,因为每个数据集(batch)中的不同donor之间除了marker外,还存在许多表达量又低生物学意义又小的基因,而这些基因用mmCorrect()也不能校正,会给后面的左图带来阻碍,因此这里选择忽略它们
进行基于MNN的校正
简单理解MNN(Mutual nearest neighbors )做了什么
想象一个情况:一个batch(A)中有一个细胞(a),然后再batch(B)中根据所选的feature表达信息找和a最相近的邻居;同样地,对batch B中的一个细胞b,也在batch A中找和它最近的邻居。像a、b细胞这种相互距离(指的是欧氏距离)最近,来自不同batch的作为一对MNN细胞
Haghverdi et al. (2018):MNN pairs represent cells from the same biological state prior to the application of a batch effect

利用MNN pair中细胞间的距离可以用来估计批次效应大小,然后差值可以作为校正批次效应的值
下面就利用mnnCorrect()函数对三个数据集(batch)进行校正批次效应,使用的基因就是chosen 得到的。下面先将三个数据集的表达量信息用logcounts提取出来,并且这个函数做了log的转换,降低了数据的维度;然后将它们放在一个列表中,并根据chosen的基因选择出来前1000个HVGs的表达量信息,是为了后面的循环使用;接着利用do.call() 对每个表达矩阵进行mnnCorrect()操作
original <- list(logcounts(sce.e5601)[chosen,],
logcounts(sce.gse81076)[chosen,],
logcounts(sce.gse85241)[chosen,])
corrected <- do.call(mnnCorrect, c(original, list(k=20, sigma=0.1)))
> str(corrected$corrected)
List of 3
$ : num [1:353, 1:2285] 0.127 0.137 0.121 0 0.113 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:353] "ENSG00000115263" "ENSG00000118271" "ENSG00000089199" "ENSG00000169903" ...
.. ..$ : chr [1:2285] "HP1502401_J13" "HP1502401_H13" "HP1502401_J14" "HP1502401_B14" ...
$ : num [1:353, 1:1292] -0.01724 -0.0062 0.01149 0.00689 0.01272 ...
$ : num [1:353, 1:2346] 0.142 0.138 0.132 0.109 0.11 ...
关于参数的解释:
k表示在定义MNN pair时,设置几个最近的邻居(nearest neighbours ),表示每个batch中每种细胞类型或状态出现的最低频率。增大这个数字,会通过增加MNN pair数量来增加矫正的精度,但是需要允许在不同细胞类型之间形成MNN pair,这一操作又会降低准确性,所以需要权衡这个数字增大k值,会提高precision,降低accuracy
sigma表示在计算批次效应时如何定义MNN pair之间共享的信息量。较大的值会共享更多信息,就像对同一批次的所有细胞都进行校正;较小的值允许跨细胞类型进行校正,可能会更准确,但会降低精度。默认值为1,比较保守的一个设定,校正不会太多,但多数情况选择小一点的值会更合适减小sigma,会增加accuracy,降低precision
这里很有必要说明两个英语词汇:
- Accuracy refers to the closeness of a measured value to a standard or known value. 和标准值比是否"准确”
- Precision refers to the closeness of two or more measurements to each other. 相互之间比是否"精确”
另外,提供的original list中各个batch的顺序是很重要的,因为是将第一个batch作为校正的参考坐标系统。一般推荐设置批次效应最大或异质性最强的批次作为对照,可以保证参考批次与其他校正批次之间有充足的MNN pair
检验校正的作用
创建一个新的SingleCellExperiment对象,将三个原始的矩阵和三个校正后的矩阵放在一起
# omat是原始矩阵,mat是校正后的
omat <- do.call(cbind, original)
mat <- do.call(cbind, corrected$corrected)
# 将mat列名去掉
colnames(mat) <- NULL
sce <- SingleCellExperiment(list(original=omat, corrected=mat))
# 用lapply对三个列表进行循环操作,求列数,为了给rep设置一个重复值
colData(sce)$Batch <- rep(c("e5601", "gse81076", "gse85241"),
lapply(corrected$corrected, ncol))
> sce
class: SingleCellExperiment
dim: 353 5923
metadata(0):
assays(2): original corrected
rownames(353): ENSG00000115263 ENSG00000118271 ... ENSG00000170348
ENSG00000251562
rowData names(0):
colnames(5923): HP1502401_J13 HP1502401_H13 ... D30.8_93 D30.8_94
colData names(1): Batch
reducedDimNames(0):
spikeNames(0):
做个t-sne图来看看
图中会显示未校正的细胞是如何根据不同批次分离的,而校正批次后细胞是混在一起的。我们希望这里能够混在一起,是为了后面的分离是真的由于生物差异
osce <- runTSNE(sce, exprs_values="original", set.seed=100)
ot <- plotTSNE(osce, colour_by="Batch") + ggtitle("Original")
csce <- runTSNE(sce, exprs_values="corrected", set.seed=100)
ct <- plotTSNE(csce, colour_by="Batch") + ggtitle("Corrected")
multiplot(ot, ct, cols=2)
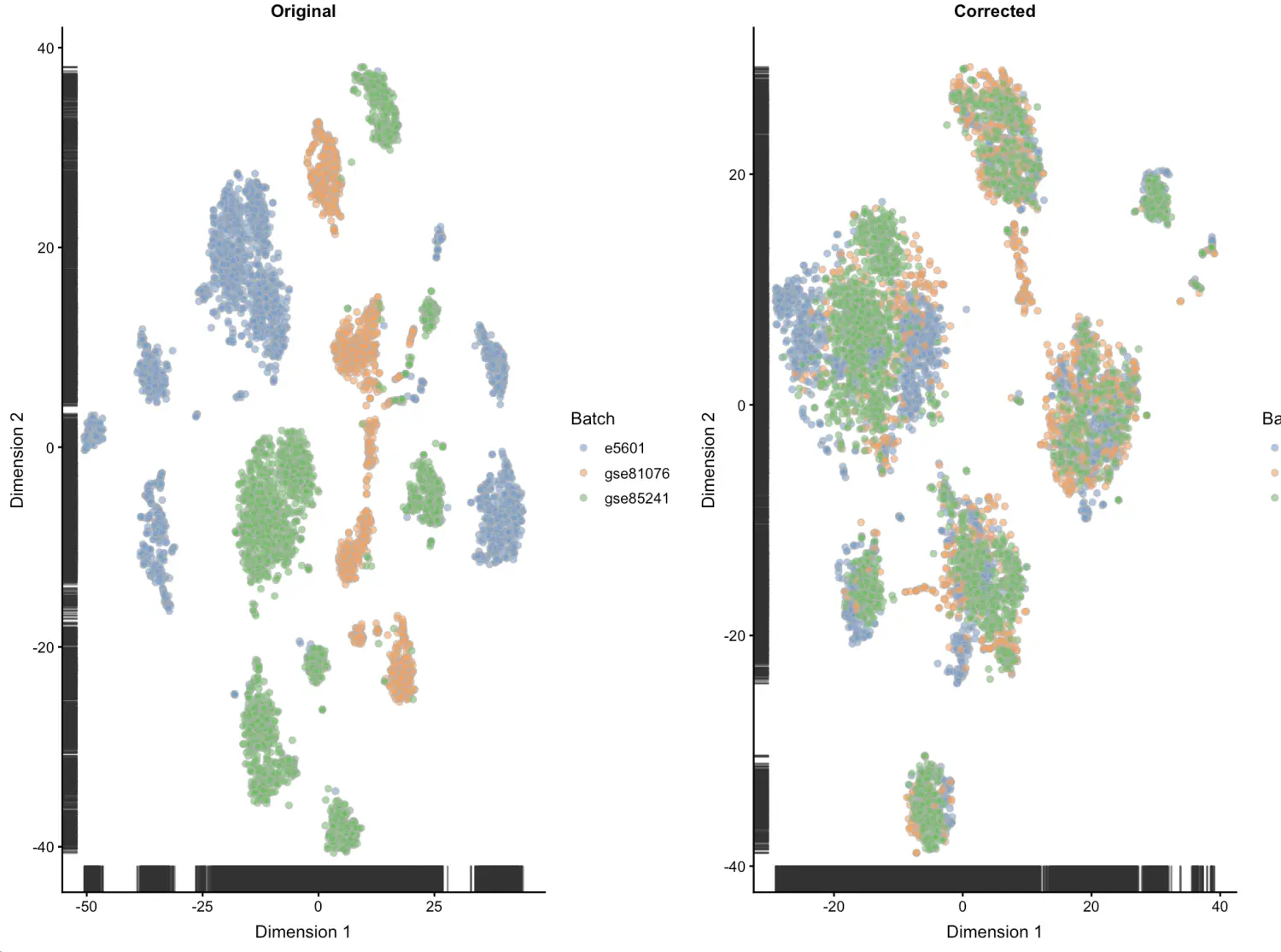
看到E-MTAB-5601这个数据集分离的最严重,推测可能其他数据集采用了UMI
然后再根据几个已知的胰腺细胞的marker基因检测一下,看看这个校正是不是能反映生物学意义。因为如果校正后虽然去除了批次效应,但如果每个群中都体现某个细胞marker基因,对后面分群也是没有意义的。
ct.gcg <- plotTSNE(csce, by_exprs_values="corrected",
colour_by="ENSG00000115263") + ggtitle("Alpha cells (GCG)")
ct.ins <- plotTSNE(csce, by_exprs_values="corrected",
colour_by="ENSG00000254647") + ggtitle("Beta cells (INS)")
ct.sst <- plotTSNE(csce, by_exprs_values="corrected",
colour_by="ENSG00000157005") + ggtitle("Delta cells (SST)")
ct.ppy <- plotTSNE(csce, by_exprs_values="corrected",
colour_by="ENSG00000108849") + ggtitle("PP cells (PPY)")
multiplot(ct.gcg, ct.ins, ct.sst, ct.ppy, cols=2)
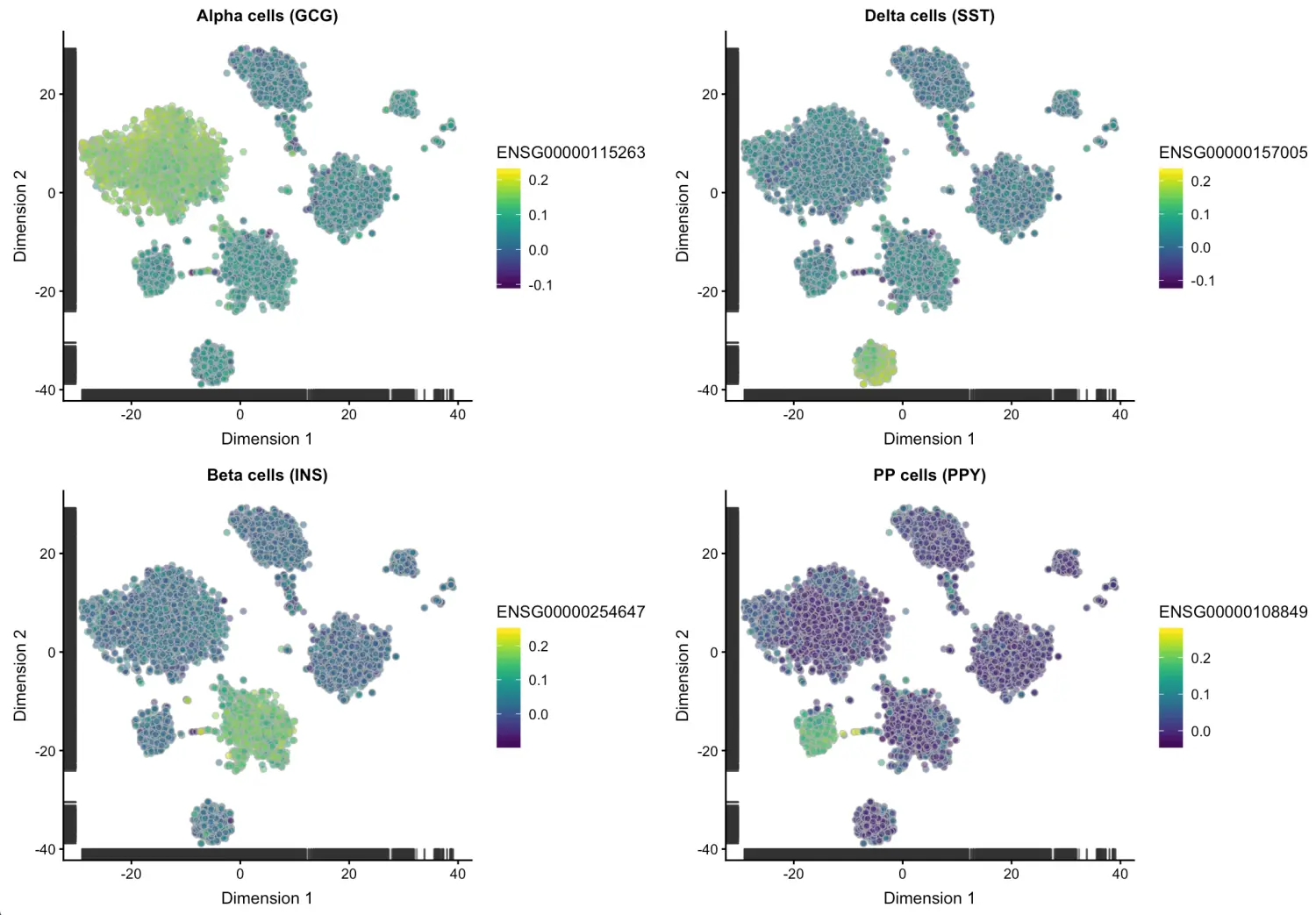
结果可以看到校正后依然可以区分细胞类型,说明既达到了减小批次效应的影响,又能不干扰后续细胞亚型的生物学鉴定