scRNA-10X免疫治疗学习笔记-7-其他R包介绍
刘小泽写于19.10.18 笔记目的:根据生信技能树的单细胞转录组课程探索10X Genomics技术相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 第二单元最后一讲第11讲:单细胞转录组的其他R包介绍
这一次的问题是:分析单细胞转录组一定要用R包吗?
之前在
10X scRNA免疫治疗学习笔记-5-差异分析及可视化 中使用monocle的plot_cell_clusters函数画出了PBMC的第4和第10群两种不同T细胞的差异。那么这个分析一定要用包装好的R包吗?不是的,即使不使用别人做的R包,自己也能利用作图原理画出来
问题的重点不在使用什么包,而是做出这个图的原理是什么
就上面这个图而言,为了分群,首先要有一个表达矩阵,还要设置分群信息(例如上面我们就明确知道要分成两群),然后还要进行PCA的线性降维和tSNE进一步非线性降维。有了这些认识,那么首先使用普通的函数来尝试一下
第一种方式: 不使用R包,使用常规函数
先加载表达矩阵和分群信息
options(warn=-1)
load('patient1.PBMC_RespD376_for_DEG.Rdata')
# 其中保存了表达矩阵(count_matrix)和分群的信息(cluster)
> count_matrix[1:4,1:4]
AAACCTGCAACGATGG.3 AAACCTGGTCTCCATC.3 AAACGGGAGCTCCTTC.3 AAAGCAATCATATCGG.3
RP11-34P13.7 0 0 0 0
FO538757.2 0 0 0 0
AP006222.2 0 0 0 0
RP4-669L17.10 0 0 0 0
> table(cluster)
cluster
4 10
636 170
根据分群因子,赋予不同颜色
# 根据分群因子,赋予不同颜色
color <- cluster
levels(color) <- rainbow(2)
> table(color)
color
#FF0000FF #00FFFFFF
636 170
对表达矩阵过滤
# 首先是对基因过滤,根据标准差将那些在细胞中的表达量没有变化的基因去掉
choosed_count <- count_matrix
choosed_count <- choosed_count[apply(choosed_count, 1, sd)>0,]
# 看到过滤了5000多个基因
# 然后选择变化差异最明显的前1000个基因
choosed_count <- choosed_count[names(head(sort(apply(choosed_count, 1, sd),decreasing = T),1000)),]
然后进行PCA分析
对行进行操作,因此需要转置,另外进行标准化
pca_out <- prcomp(t(choosed_count),scale. = T)
library(ggfortify)
autoplot(pca_out, col=color) +theme_classic()+ggtitle('PCA plot')
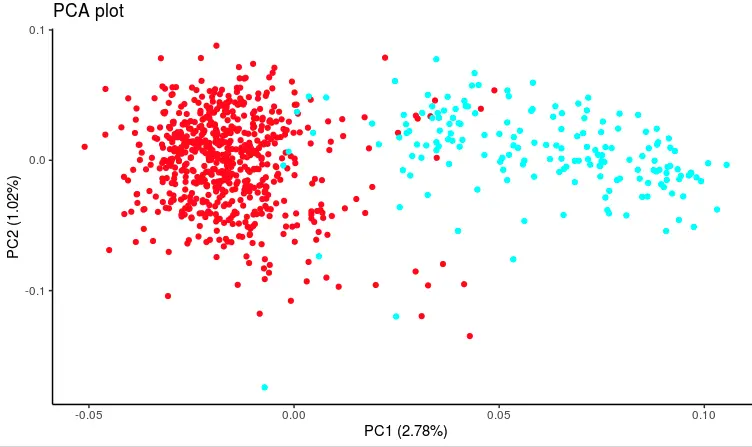
PCA的全部结果可以通过str(pca_out)了解,其中坐标的信息在pca_out$x
> pca_out$x[1:3,1:3]
PC1 PC2 PC3
AAACCTGCAACGATGG.3 -1.4940046 6.707691 -0.7655250
AAACCTGGTCTCCATC.3 0.1403445 3.239395 0.2672606
AAACGGGAGCTCCTTC.3 -1.9968613 3.924992 -0.4669826
再用tSNE继续降维
library(Rtsne)
# 利用PCA的前5个主成分
tsne_out <- Rtsne(pca_out$x[,1:6], perplexity = 10,
pca = F, max_iter = 2000,
verbose = T)
# tSNE的坐标结果保存在tsne_out$Y中
tsnes_cord <- tsne_out$Y
# ggplot作图
colnames(tsnes_cord) <- c('tSNE1','tSNE2')
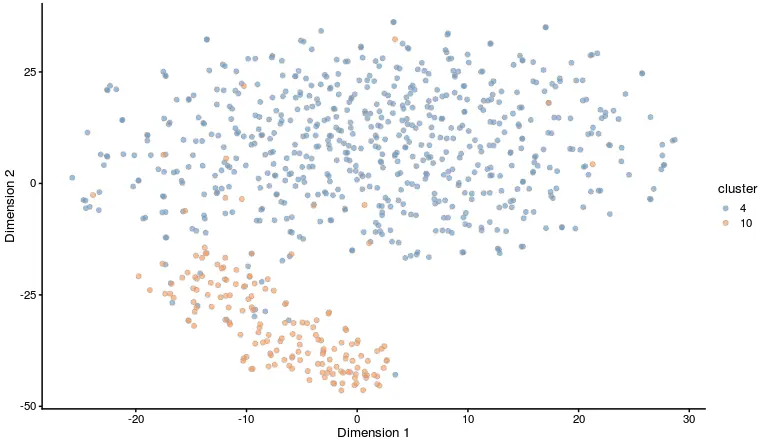
ggplot(tsnes_cord, aes(x=tSNE1, y = tSNE2)) + geom_point(col=color) + theme_classic()+ggtitle('tSNE plot')
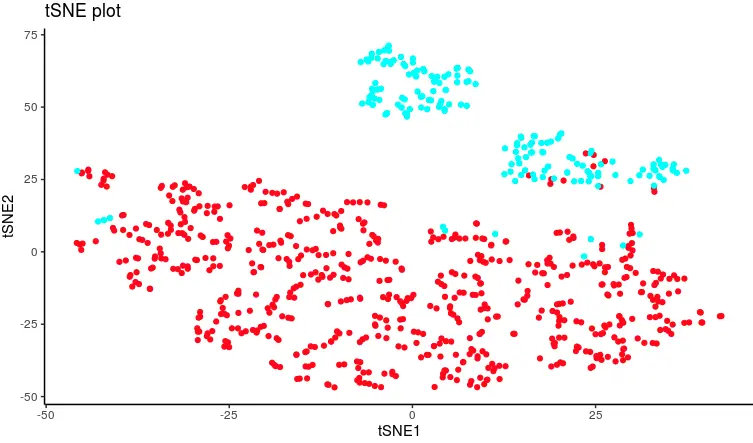
第二种方式:使用Seurat
Seurat V2
PBMC <- CreateSeuratObject(count_matrix,
min.cells = 1, min.features = 0,
project = '10x_PBMC')
PBMC <- AddMetaData(object = PBMC, metadata = apply(count_matrix, 2, sum), col.name = 'nUMI_raw')
PBMC <- AddMetaData(object = PBMC, metadata = cluster, col.name = 'cluster')
VlnPlot(PBMC, features.plot = c("nUMI_raw", "nGene"),
group.by = 'cluster',nCol = 2)
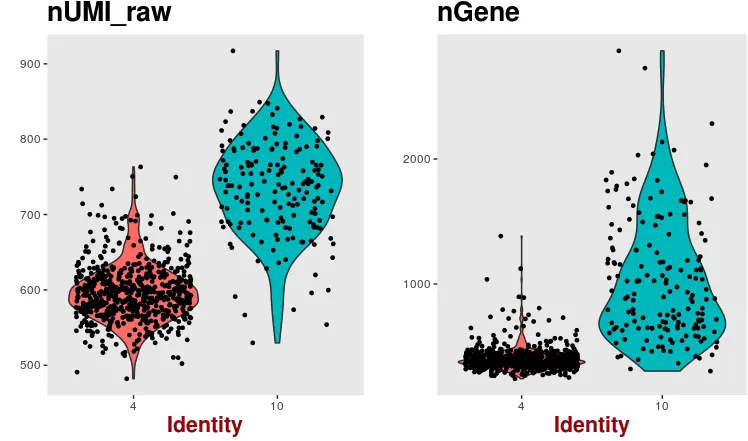
看到第10群细胞的基因数和UMI数比第4群多很多,也就是说它们本身的“起跑线”就有差异,那么最后的PCA结果差异难道是由于基因和UMI数量导致的?
那么如果我们接下来进行校正,会发生什么?
PBMC <- ScaleData(PBMC, vars.to.regress = c("nUMI_raw", "nGene"),
model.use = 'linear',
use.umi = F)
# 这个FindVariableGenes其实和前面的找top1000 sd基因目的一样,就是为了增加分析的有效性,认为高变化的基因存储的信息更值得关注
PBMC <- FindVariableGenes(object = PBMC, mean.function = ExpMean,
dispersion.function = LogVMR,
x.low.cutoff = 0.0125, x.high.cutoff = 3,
y.cutoff = 0.5)
length(PBMC@var.genes)
PBMC <- RunPCA(object = PBMC, pc.genes = PBMC@var.genes)
PBMC <- RunTSNE(object = PBMC, dims.use = 1:20)
TSNEPlot(PBMC, group.by='cluster')
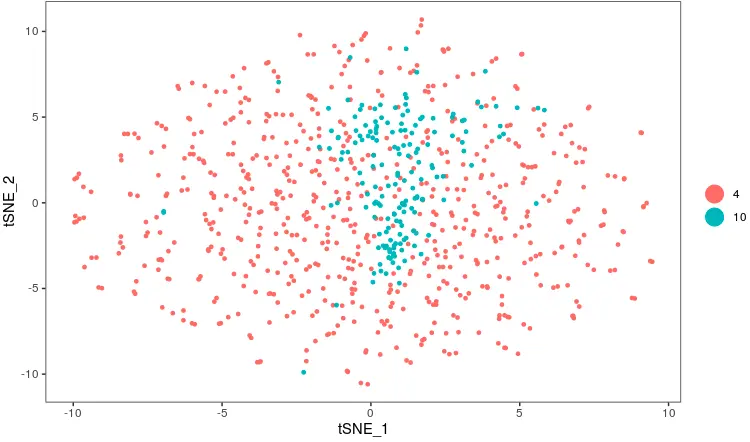
这次结果发现,这两群就分不开了。从这里也反映出一些问题:本文的这个热图真的是由于生物学因素导致的吗?
猜想:可能这两群细胞本身表达的基因数量就不同,就是有一些基因在这群细胞表达,在那一群不表达
Seurat V3
PBMC_V3 <- CreateSeuratObject(counts = count_matrix,
min.cells = 1,
min.features = 0,
project = "10x_PBMC_V3")
PBMC_V3 <- AddMetaData(object = PBMC_V3, metadata = apply(count_matrix, 2, sum), col.name = 'nUMI_raw')
PBMC_V3 <- AddMetaData(object = PBMC_V3, metadata = cluster, col.name = 'cluster')
VlnPlot(PBMC_V3, features = c("nUMI_raw", "nFeature_RNA"), ncol = 2,group.by = 'cluster')
# 同样也进行校正一下
PBMC_V3 <- ScaleData(object = PBMC_V3, vars.to.regress = c("nUMI_raw", "nFeature_RNA"),
model.use = 'linear', use.umi = FALSE)
PBMC_V3 <- FindVariableFeatures(object = PBMC_V3, mean.function = ExpMean,
dispersion.function = LogVMR,
mean.cutoff = c(0.0125,3),
dispersion.cutoff = c(0.5,Inf))
length(VariableFeatures(object = PBMC_V3))
PBMC_V3 <- RunPCA(PBMC_V3, features = VariableFeatures(object = PBMC_V3))
pbmc_tsne <- RunTSNE(PBMC_V3, dims = 1:20)
DimPlot(pbmc_tsne, reduction = "tsne",group.by='cluster')
第三种方式:使用monocle
library(monocle)
# 1.表达矩阵
expr_matrix <- as.matrix(count_matrix)
# 2.细胞信息
sample_ann <- data.frame(cells=names(count_matrix),
cellType=cluster)
rownames(sample_ann)<- names(count_matrix)
# 3.基因信息
gene_ann <- as.data.frame(rownames(count_matrix))
rownames(gene_ann)<- rownames(count_matrix)
colnames(gene_ann)<- "genes"
# 然后转换为AnnotatedDataFrame对象
pd <- new("AnnotatedDataFrame",
data=sample_ann)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
# 最后构建CDS对象
sc_cds <- newCellDataSet(
expr_matrix,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=0.5)
cds=sc_cds
cds <- detectGenes(cds, min_expr = 1)
expressed_genes <- row.names(subset(cds@featureData@data,
num_cells_expressed >= 1))
length(expressed_genes)
cds <- cds[expressed_genes,]
cds <- estimateSizeFactors(cds)
cds <- estimateDispersions(cds)
disp_table <- dispersionTable(cds) # 挑有差异的
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1) # 挑表达量不太低的
cds <- setOrderingFilter(cds, unsup_clustering_genes$gene_id) # 准备聚类基因名单
# 进行降维
cds <- reduceDimension(cds, max_components = 2, num_dim = 6,
reduction_method = 'tSNE', verbose = T)
# 进行聚类
cds <- clusterCells(cds, num_clusters = 4)
# Distance cutoff calculated to 1.812595
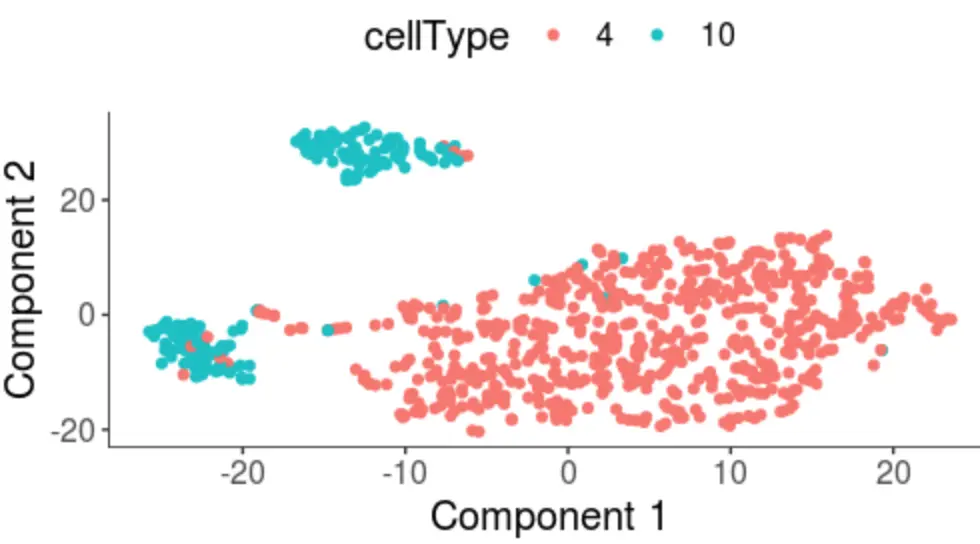
plot_cell_clusters(cds, 1, 2, color = "cellType")
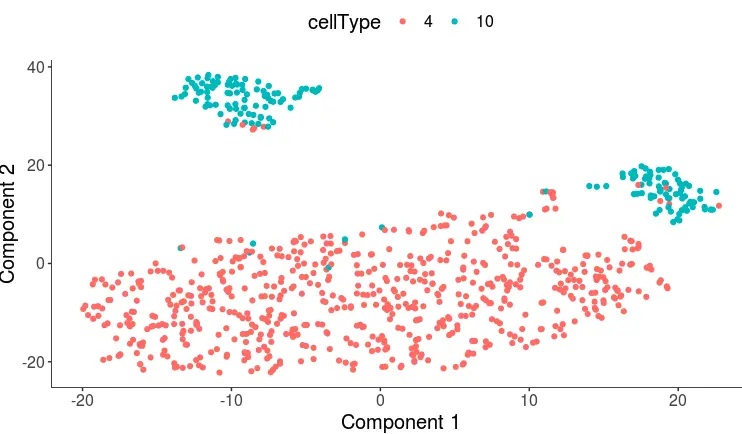
第四种方式:使用scater
suppressMessages(library(scater))
## 创建 scater 要求的对象
# 在导入对象之前,最好是将表达量数据存为矩阵
sce <- SingleCellExperiment(
assays = list(counts = as.matrix(count_matrix)),
colData = data.frame(cluster)
)
# 预处理
exprs(sce) <- log2(calculateCPM(sce ) + 1)
# 降维
sce <- runPCA(sce)
plotReducedDim(sce, use_dimred = "PCA",
colour_by= "cluster")
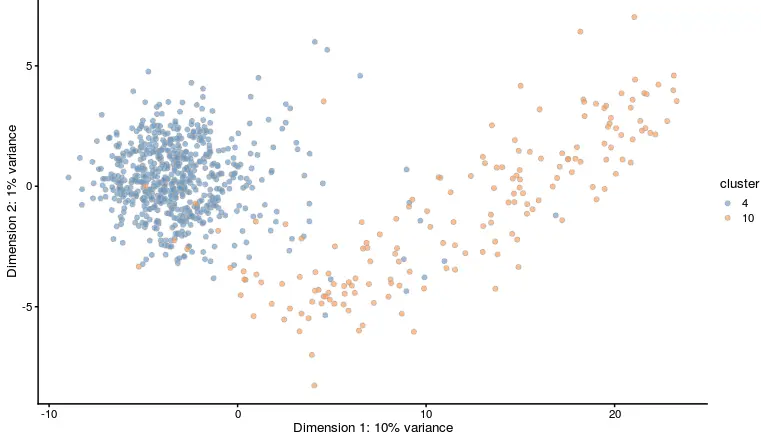
set.seed(1000)
sce <- runTSNE(sce, perplexity=10)
plotTSNE(sce,
colour_by= "cluster")