scRNA-10X免疫治疗学习笔记-6-marker基因的表达量可视化
刘小泽写于19.10.18 笔记目的:根据生信技能树的单细胞转录组课程探索10X Genomics技术相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 第二单元第10讲:marker基因的表达量可视化
前言
这次的任务是模仿原文的:
肿瘤组织的差异分析(Fig. 4a、c)
下载数据
之前的分析都是基于第一个病人的PBMC,这次将基于这位病人的tumor: GSE117988_raw.expMatrix_Tumor.csv.gz
start_time <- Sys.time()
raw_dataTumor <- read.csv('./GSE117988_raw.expMatrix_Tumor.csv.gz', header = TRUE, row.names = 1)
end_time <- Sys.time()
end_time - start_time
# Time difference of 47.00589 secs
dim(raw_dataTumor) # 21861基因,7431细胞- already filtered
注意:之前PBMC包含了四个时间点的样本,这里的Tumor包含了2个样本(治疗之前Pre和复发AR)
常规流程
step1: 归一化
dataTumor <- log2(1 + sweep(raw_dataTumor, 2, median(colSums(raw_dataTumor))/colSums(raw_dataTumor), '*'))
> head(colnames(dataTumor))
[1] "AAACCTGAGGATGTAT.1" "AAACCTGCAGCGATCC.1" "AAACCTGGTACGAAAT.1" "AAACGGGAGCTGGAAC.1" "AAACGGGAGGAGTTGC.1"
[6] "AAACGGGAGTTTAGGA.1"
step2: 自定义划分细胞类型
cellTypes <- sapply(colnames(dataTumor), function(x) ExtractField(x, 2, '[.]'))
cellTypes <-ifelse(cellTypes == '1', 'Tumor_Before', 'Tumor_AcquiredResistance')
> table(cellTypes)
cellTypes
Tumor_AcquiredResistance Tumor_Before
5188 2243
step3: 表达矩阵质控
# 第一点:基因在多少细胞表达
> fivenum(apply(dataTumor,1,function(x) sum(x>0) ))
VP2 GPRIN2 EML3 ZNF140 RPLP1
1 8 103 566 7431
# 第二点:细胞中有多少表达的基因
> fivenum(apply(dataTumor,2,function(x) sum(x>0) ))
GGAACTTAGGAATCGC.1 TACGGTACAAGCCGCT.2 CCTACCAAGCGTGAGT.1 TGAGCCGAGACTAGAT.2 GATCGTAGTCATATGC.2
192.0 1059.0 1380.0 1971.5 5888.0
看到大部分基因在500多个细胞表达,细胞平均能表达1000个基因以上
step4: 创建Seurat对象
tumor <- CreateSeuratObject(dataTumor,
min.cells = 1, min.features = 0, project = '10x_Tumor')
> tumor
An object of class seurat in project 10x_Tumor
21861 genes across 7431 samples.
step5: 添加metadata (nUMI 和 细胞类型)
tumor <- AddMetaData(object = tumor, metadata = apply(raw_dataTumor, 2, sum), col.name = 'nUMI_raw')
tumor <- AddMetaData(object = tumor, metadata = cellTypes, col.name = 'cellTypes')
step6: 聚类标准流程
start_time <- Sys.time()
tumor <- ScaleData(object = tumor, vars.to.regress = c('nUMI_raw'), model.use = 'linear', use.umi = FALSE)
tumor <- FindVariableGenes(object = tumor, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5)
tumor <- RunPCA(object = tumor, pc.genes = tumor@var.genes)
tumor <- RunTSNE(object = tumor, dims.use = 1:10, perplexity = 25)
end_time <- Sys.time()
end_time - start_time
# Time difference of 3.324982 mins
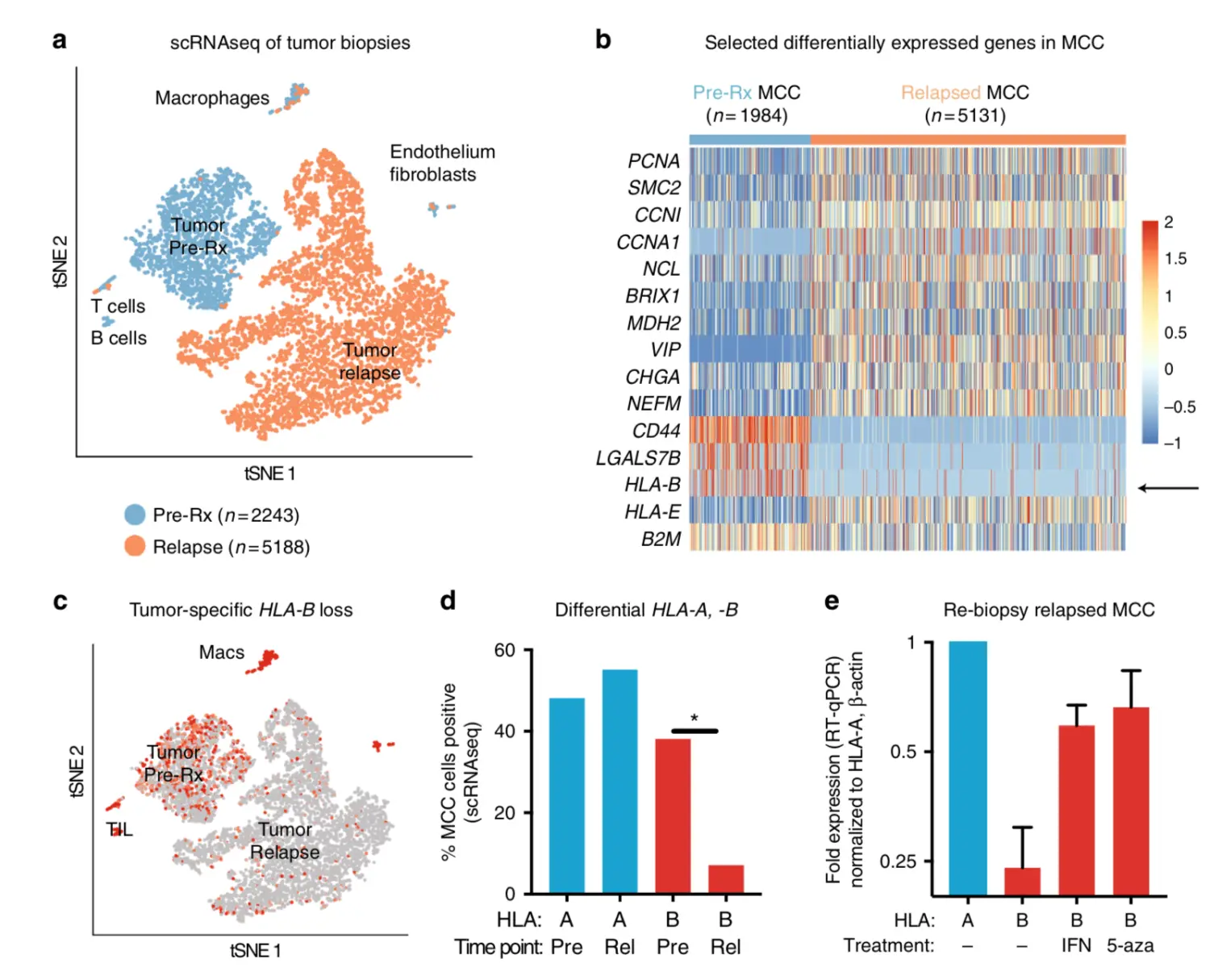
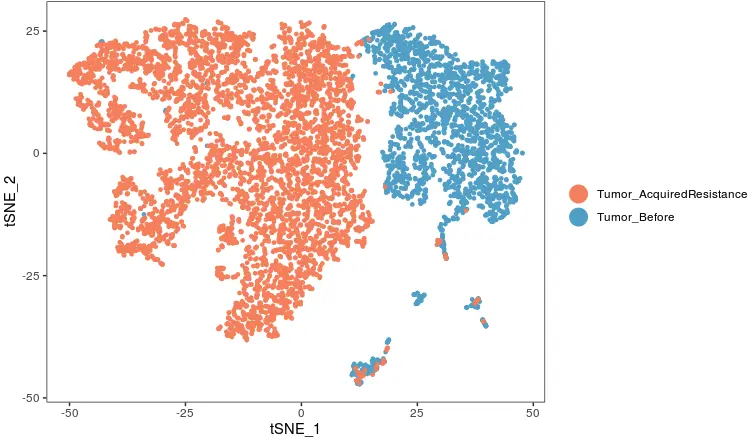
TSNEPlot(tumor, group.by = 'cellTypes', colors.use = c('#EF8A62', '#67A9CF'))
save(tumor,file = 'patient1.Tumor.V2.output.Rdata') # 3.6Gb 大小
接着进行基因可视化
rm(list = ls())
options(warn=-1)
start_time <- Sys.time()
load('patient1.Tumor.V2.output.Rdata')
end_time <- Sys.time()
end_time - start_time
# Time difference of 19.83097 secs
取出log归一化后的表达矩阵
count_matrix=tumor@data
> count_matrix[1:4,1:4]
AAACCTGAGGATGTAT.1 AAACCTGCAGCGATCC.1 AAACCTGGTACGAAAT.1 AAACGGGAGCTGGAAC.1
VP2 0.0000000 0 0 0
largeTAntigen 0.9670525 0 0 0
smallTAntigen 0.0000000 0 0 0
RP11-34P13.7 0.0000000 0 0 0
取出细胞分群信息
cluster=tumor@meta.data$cellTypes
> table(cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
5188 2243
提取基因信息
文章主要探索了治疗前和复发后的HLA-A和HLA-B的变化,于是我们先看看有没有这两个基因
allGenes = row.names(tumor@raw.data)
> allGenes[grep('HLA',allGenes)]
[1] "HHLA3" "HLA-F" "HLA-G" "HLA-A" "HLA-E" "HLA-C" "HLA-B" "HLA-DRA" "HLA-DRB5"
[10] "HLA-DRB1" "HLA-DQA1" "HLA-DQB1" "HLA-DQA2" "HLA-DQB2" "HLA-DOB" "HLA-DMB" "HLA-DMA" "HLA-DOA"
[19] "HLA-DPA1" "HLA-DPB1"
对HLA-A操作
FeaturePlot(object = tumor,
features.plot ='HLA-A',
cols.use = c("grey", "blue"),
reduction.use = "tsne")
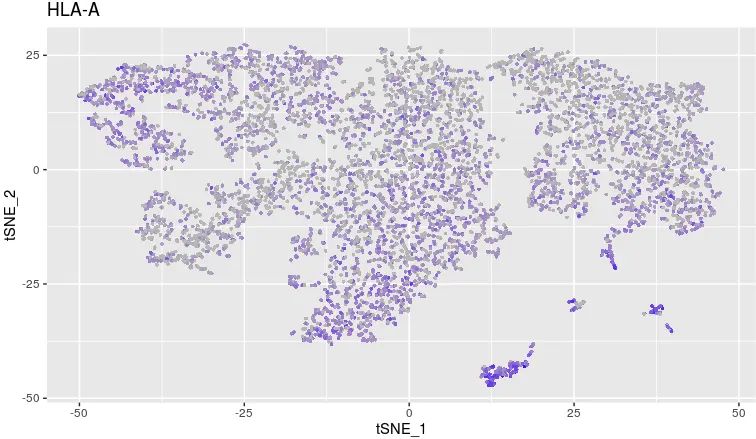
看HLA-A表达量
> table(count_matrix['HLA-A',]>0, cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
FALSE 2282 1057
TRUE 2906 1186
对HLA-B操作
FeaturePlot(object = tumor,
features.plot ='HLA-B',
cols.use = c("grey", "blue"),
reduction.use = "tsne")
可以看到治疗前HLA-A基因有1186个表达,1057个不表达;复发后这个基因表达和不表达的数量也相近
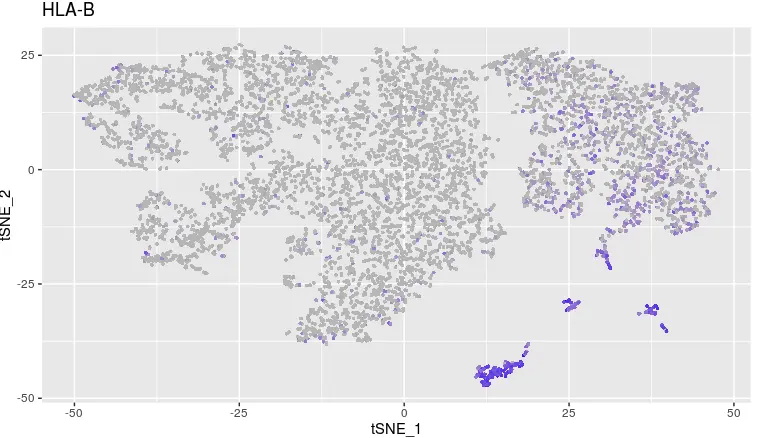
> table(count_matrix['HLA-B',]>0, cluster)
cluster
Tumor_AcquiredResistance Tumor_Before
FALSE 4794 1258
TRUE 394 985
对HLA-B来讲,不管是治疗前还是复发后,它的表达和不表达差异就很明显。另外从治疗前到复发,这个基因的表达数量的变化更显著
小问题:HLA基因这么多,我们怎么找到全部的具有相似模式的基因?
所谓相似表达模式,就是像HLA-B基因一样,在一个群表达很多,另一个群表达很少,通过 卡方检验就能看出区别
> chisq.test(table(count_matrix['HLA-A',]>0, cluster))
Pearson's Chi-squared test with Yates' continuity correction
data: table(count_matrix["HLA-A", ] > 0, cluster)
X-squared = 6.1069, df = 1, p-value = 0.01347
> chisq.test(table(count_matrix['HLA-B',]>0, cluster))
Pearson's Chi-squared test with Yates' continuity correction
data: table(count_matrix["HLA-B", ] > 0, cluster)
X-squared = 1364.4, df = 1, p-value < 2.2e-16
看p值,HLA-B显著性相比HLA-A就非常强,我们就是要挑出和HLA-B类似的,也就是极显著的,设定p值的阈值为0.01
HLA_genes <- allGenes[grep('HLA',allGenes)]
# 将输出结果保存在向量中
HLA_result <- c()
for (gene in HLA_genes) {
tmp <- chisq.test(table(count_matrix[gene,]>0, cluster))
if (tmp$p.value<0.01) {
HLA_result[gene] <- gene
}
}
> names(HLA_result)
[1] "HLA-F" "HLA-E" "HLA-C" "HLA-B" "HLA-DRA" "HLA-DRB5" "HLA-DRB1" "HLA-DQA1" "HLA-DQB1"
[10] "HLA-DQA2" "HLA-DMB" "HLA-DMA" "HLA-DOA" "HLA-DPA1" "HLA-DPB1"
> length(HLA_result)
[1] 15