scRNA-10X免疫治疗学习笔记-3-走Seurat标准流程
刘小泽写于19.10.15 笔记目的:根据生信技能树的单细胞转录组课程探索10X Genomics技术相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 第二单元第7讲:走Seurat标准流程【文章结构总-分-总,结尾有完整的代码,熟悉者前面可以跳过,去看后面8min完成的代码】
前言
前面介绍了自己利用cellranger count的结果进行seurat分析,但是整合数据方面做得还是不如原作者优秀,虽然我们不知道他们是如何处理的,但还是可以继续向下进行,而且这一次将会使用他们合并好的数据
从下载GEO开始
https://www.ncbi.nlm.nih.gov//geo/query/acc.cgi?acc=GSE117988
下载: GSE117988_raw.expMatrix_PBMC.csv.gz
rm(list = ls())
options(warn=-1)
suppressMessages(library(Seurat))
# 读取表达矩阵
start_time <- Sys.time()
raw_dataPBMC <- read.csv('./GSE117988_raw.expMatrix_PBMC.csv.gz', header = TRUE, row.names = 1)
end_time <- Sys.time()
end_time - start_time
#Time difference of 1.486653 mins
> dim(raw_dataPBMC)
[1] 17712 12874
# 得到17712基因,12874细胞
复现作者的分群结果
下载后怎么进行归一化、时间点划分、创建对象、分群其实作者也已经给了答案:https://www.researchgate.net/publication/328016998_Supplementary_Material_6/data/5bb2eac9299bf13e605a0a74/41467-2018-6300-MOESM6-ESM.txt
首先对文库大小进行一个归一化
dataPBMC <- log2(1 + sweep(raw_dataPBMC, 2, median(colSums(raw_dataPBMC))/colSums(raw_dataPBMC), '*'))
这个sweep函数很有趣
它可以在apply的统一操作基础上,增加个性化服务(比如对每行/列使用不同的数值进行计算,而且可以指定计算方法)
例如,想要将1-3行分别减去对应的行号1-3,就可以这样:
> test <- matrix(1:12,ncol = 4,byrow = T)
> test
[,1] [,2] [,3] [,4]
[1,] 1 2 3 4
[2,] 5 6 7 8
[3,] 9 10 11 12
> sweep(test,1,c(1,2,3),"-")
[,1] [,2] [,3] [,4]
[1,] 0 1 2 3
[2,] 3 4 5 6
[3,] 6 7 8 9
- 第一个位置
test这里需要是矩阵或数据框; - 第二个位置
1和2选一个,原理和apply一样 - 第三个位置是要操作的向量,如果要对行操作,那么这个向量长度就要和行数一样
- 第四个位置是计算符,比如:
+ - * / < >等
于是就能懂了这里的操作:先求每个细胞文库的总大小,然后用它的中位数除以总大小得到一个小数,然后按列乘以这个小数就相当于对文库进行了归一化,将文库本身的大小差异置之度外
自定义划分时间点
看看现在的细胞命名,其实我们看完文章是知道作者用了四个时间点的,当然,作者在做实验过程中,也会有自己的一套方法对不同时间点的细胞进行命名。比如作者这里的想法就是:
> head(colnames(dataPBMC))
[1] "AAACCTGAGCGAAGGG.1" "AAACCTGAGGTCATCT.1" "AAACCTGAGTCCTCCT.1"
[4] "AAACCTGCACCAGCAC.1" "AAACCTGGTAACGTTC.1" "AAACCTGGTAAGGATT.1"
> tail(colnames(dataPBMC))
[1] "TTTGTCAAGCGAGAAA.4" "TTTGTCAAGGAATTAC.4" "TTTGTCAAGTGCGTGA.4"
[4] "TTTGTCACACGAGGTA.4" "TTTGTCATCATTGCGA.4" "TTTGTCATCCACGCAG.4"
看到每个细胞barcode后面都有一个点号,然后接着数字1-4来区分四个时间点,并且是按照时间顺序来的,它们的对应顺序就是:
1 => PBMC_Pre
2 => PBMC_EarlyD27
3 => PBMC_RespD376
4 => PBMC_ARD614
作者的想法是:将数字提取出来,然后分别对应到具体时期名称(利用if else结构)
# 怎么提取?
# 作者利用的是Seurat V2的ExtractField函数
# for Seurat V2
timePoints <- sapply(colnames(dataPBMC),
function(x) ExtractField(x, 2, '[.]'))
# 但如果使用V3,就要用常规方法strsplit,并且注意点号是正则匹配符,需要用\\来转义
timePoints <- sapply(colnames(dataPBMC), function(x) unlist(strsplit(x, "\\."))[2])
> table(timePoints)
timePoints
1 2 3 4
2082 1592 4684 4516
完成对应
timePoints <-ifelse(timePoints == '1', 'PBMC_Pre',
ifelse(timePoints == '2', 'PBMC_EarlyD27',
ifelse(timePoints == '3', 'PBMC_RespD376', 'PBMC_ARD614')))
> table(timePoints)
timePoints
PBMC_ARD614 PBMC_EarlyD27 PBMC_Pre PBMC_RespD376
4516 1592 2082 4684
对表达矩阵进行质控
主要看两点:
# 第一点:基因在多少细胞表达
fivenum(apply(dataPBMC,1,function(x) sum(x>0) ))
# RP4-669L17.10 LAMB3 NAT10 AC093673.5 RPL21
# 1 6 50 207 12102
boxplot(apply(dataPBMC,1,function(x) sum(x>0) ))
# 第二点:细胞中有多少表达的基因
fivenum(apply(dataPBMC,2,function(x) sum(x>0) ))
# CAAGAAATCGATCCCT.2 GGCCGATTCCAGAGGA.3 TCAACGAAGAGCTGGT.3
# 10 321 395
# TGCCAAAGTCTGCGGT.4 TCTGGAATCTATCGCC.3
# 481 2865
hist(apply(dataPBMC,2,function(x) sum(x>0) ))
看到:在一万多个基因中,75%的基因只在200多个细胞中有表达;而在一万多个细胞中,75%的细胞也只表达不到500个基因。按照常理,10X的数据应该能做到平均表达800个基因
创建Seurat对象
# Seurat V2
PBMC <- CreateSeuratObject(raw.data = dataPBMC,
min.cells = 1, min.genes = 0, project = '10x_PBMC')
# Seurat V3
PBMC <- CreateSeuratObject(dataPBMC,
min.cells = 1, min.features = 0, project = '10x_PBMC')
> PBMC
An object of class Seurat
17712 features across 12874 samples within 1 assay
Active assay: RNA (17712 features)
添加metadata
# Seurat V2 使用AddMetaData
# 3.0版本可以直接使用 object$name <- vector,当然也可以用AddMetaData
PBMC <- AddMetaData(object = PBMC,
metadata = apply(raw_dataPBMC, 2, sum),
col.name = 'nUMI_raw')
PBMC <- AddMetaData(object = PBMC, metadata = timePoints, col.name = 'TimePoints')
聚类标准流程
标准化=》找高变异基因=》根据这些基因进行PCA降维=》根据PCA结果找分群=》TSNE降维=》可视化
# Seurat V2
PBMC <- ScaleData(object = PBMC, vars.to.regress = c('nUMI_raw'), model.use = 'linear', use.umi = FALSE)
PBMC <- FindVariableGenes(object = PBMC, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5)
PBMC <- RunPCA(object = PBMC, pc.genes = PBMC@var.genes)
PBMC <- FindClusters(object = PBMC, reduction.type = "pca", dims.use = 1:10, resolution = 1, k.param = 35, save.SNN = TRUE) # 13 clusters
PBMC <- RunTSNE(object = PBMC, dims.use = 1:10)
TSNEPlot(PBMC, colors.use = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black'))
如果使用Seurat V3,会发生一些变化
3.0版本将
FindVariableGenes换为FindVariableFeatures,另外将原来的cutoff进行整合,x轴统一归到mean.cutoff中,y轴归到dispersion.cutoff中PBMC <- FindVariableFeatures(object = PBMC, mean.function = ExpMean, dispersion.function = LogVMR, mean.cutoff = c(0.0125,3), dispersion.cutoff = c(0.5,Inf))3.0版本将
FindClusters拆分为FindNeighbors和FindClusters,而版本2只有一个函数FindClustersPBMC <- FindNeighbors(PBMC, reduction = "pca", dims = 1:10, k.param = 35) PBMC <- FindClusters(object = PBMC, resolution = 0.9, verbose=F)3.0版本在最后的可视化上可以使用
DimPlot、TSNEPlot,颜色参数变成了colsDimPlot(PBMC, cols = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black'))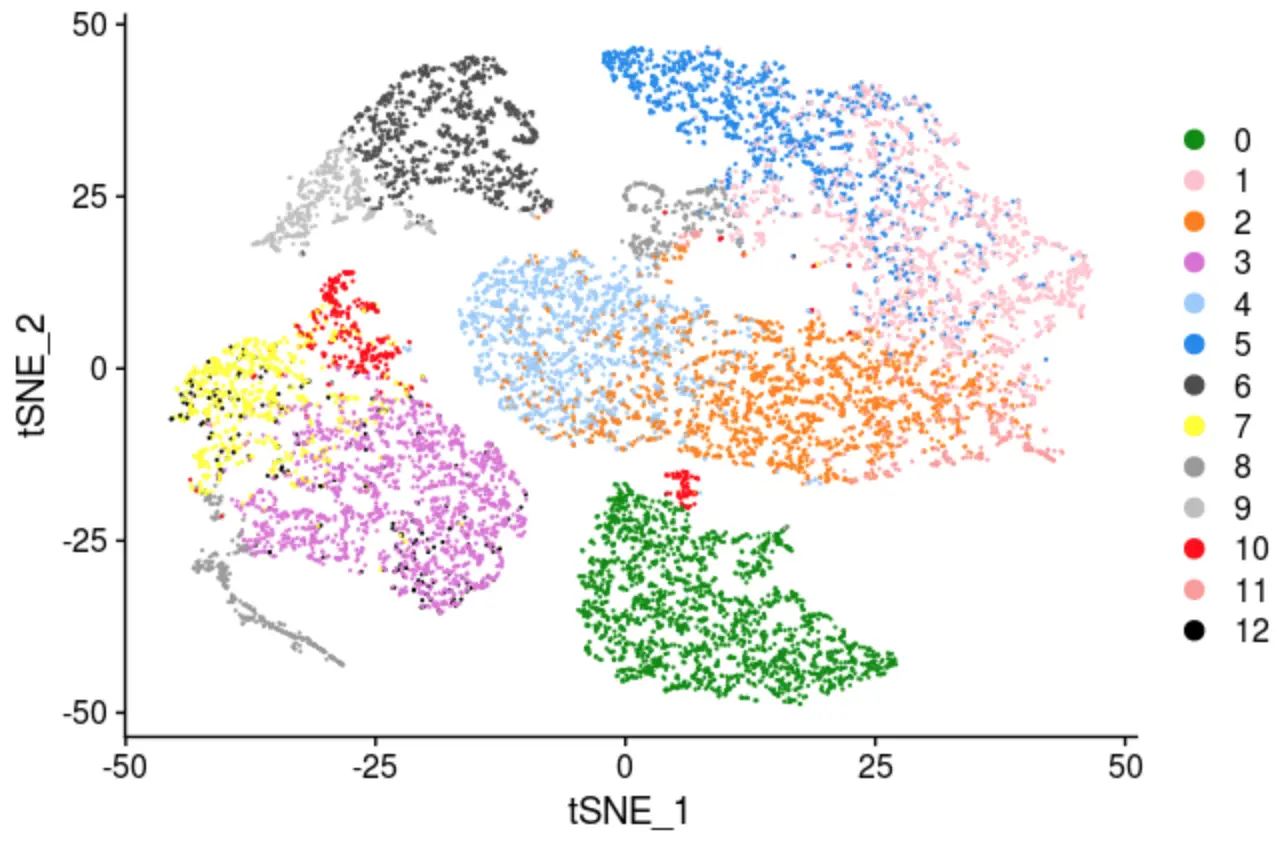
【注意】如果参数使用不正确,DimPlot或TSNEPlot会调用默认颜色设置,例如使用参数
colors或colors.use都是V3不识别的,因此它不会按照我们的颜色操作,而是生成类似这种:
保存对象
save(PBMC,file = 'patient1.PBMC.output.Rdata')
# 结果有1.9G
附加 一个问题:Seurat2、3得到的结果差别大吗?
就看最后的TSNE聚类图,上面用Seurat3操作了一遍,虽然使用了和作者一样的数据,但结果和原文还是差别很大。
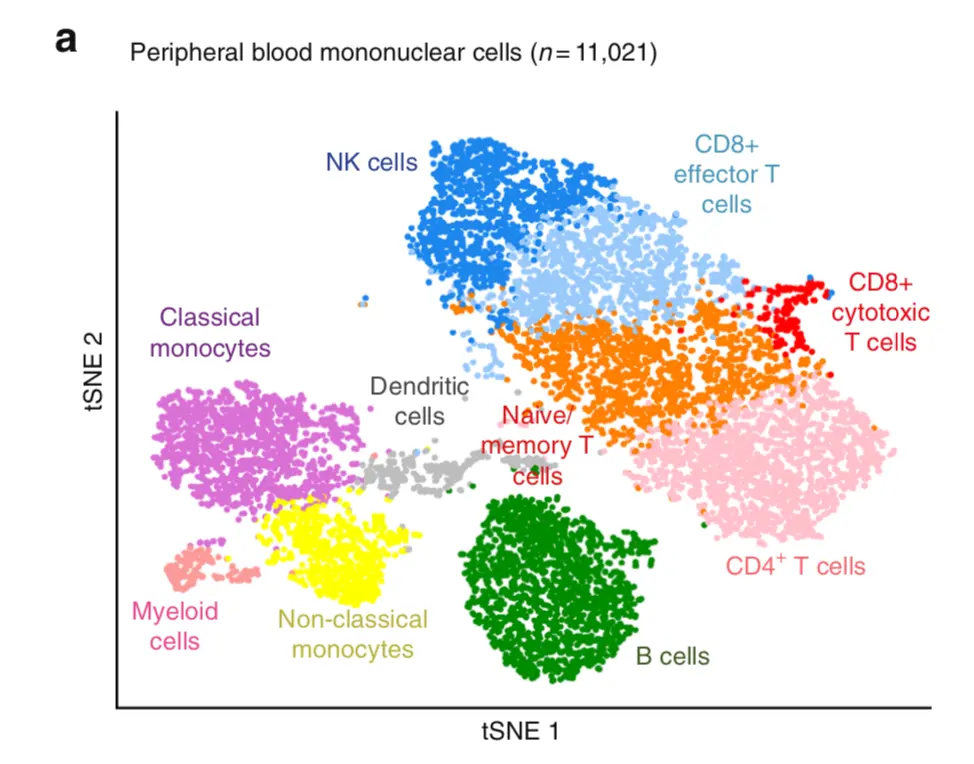
那么原因真的是由于版本引起的吗?使用作者的V2会不会好一些?
下面进行V2测试
8min跑一遍
rm(list = ls())
options(warn=-1)
suppressMessages(library(Seurat))
#############################
# 读取表达矩阵
#############################
start_time <- Sys.time()
raw_dataPBMC <- read.csv('./GSE117988_raw.expMatrix_PBMC.csv.gz', header = TRUE, row.names = 1)
## step1: 归一化
dataPBMC <- log2(1 + sweep(raw_dataPBMC, 2,
median(colSums(raw_dataPBMC))/colSums(raw_dataPBMC), '*')) # Normalization
## step2: 自定义划分时间点
# 作者利用的是Seurat V2的ExtractField函数
# for Seurat V2
timePoints <- sapply(colnames(dataPBMC),
function(x) ExtractField(x, 2, '[.]'))
timePoints <-ifelse(timePoints == '1', 'PBMC_Pre',
ifelse(timePoints == '2', 'PBMC_EarlyD27',
ifelse(timePoints == '3', 'PBMC_RespD376', 'PBMC_ARD614')))
## step3: 表达矩阵质控
# 第一点:基因在多少细胞表达
fivenum(apply(dataPBMC,1,function(x) sum(x>0) ))
# 第二点:细胞中有多少表达的基因
fivenum(apply(dataPBMC,2,function(x) sum(x>0) ))
## step4: 创建Seurat对象
PBMC <- CreateSeuratObject(dataPBMC,
min.cells = 1, min.features = 0, project = '10x_PBMC')
PBMC # 17,712 genes and 12,874 cells
## step5: 添加metadata (nUMI and timePoints)
PBMC <- AddMetaData(object = PBMC, metadata = apply(raw_dataPBMC, 2, sum), col.name = 'nUMI_raw')
PBMC <- AddMetaData(object = PBMC, metadata = timePoints, col.name = 'TimePoints')
## step6: 聚类标准流程
# Seurat V2
PBMC <- ScaleData(object = PBMC, vars.to.regress = c('nUMI_raw'), model.use = 'linear', use.umi = FALSE)
PBMC <- FindVariableGenes(object = PBMC, mean.function = ExpMean, dispersion.function = LogVMR, x.low.cutoff = 0.0125, x.high.cutoff = 3, y.cutoff = 0.5)
PBMC <- RunPCA(object = PBMC, pc.genes = PBMC@var.genes)
PBMC <- FindClusters(object = PBMC, reduction.type = "pca", dims.use = 1:10, resolution = 1, k.param = 35, save.SNN = TRUE) # 13 clusters
PBMC <- RunTSNE(object = PBMC, dims.use = 1:10)
TSNEPlot(PBMC, colors.use = c('green4', 'pink', '#FF7F00', 'orchid', '#99c9fb', 'dodgerblue2', 'grey30', 'yellow', 'grey60', 'grey', 'red', '#FB9A99', 'black'))
end_time <- Sys.time()
end_time - start_time
# Time difference of 8.085459 mins
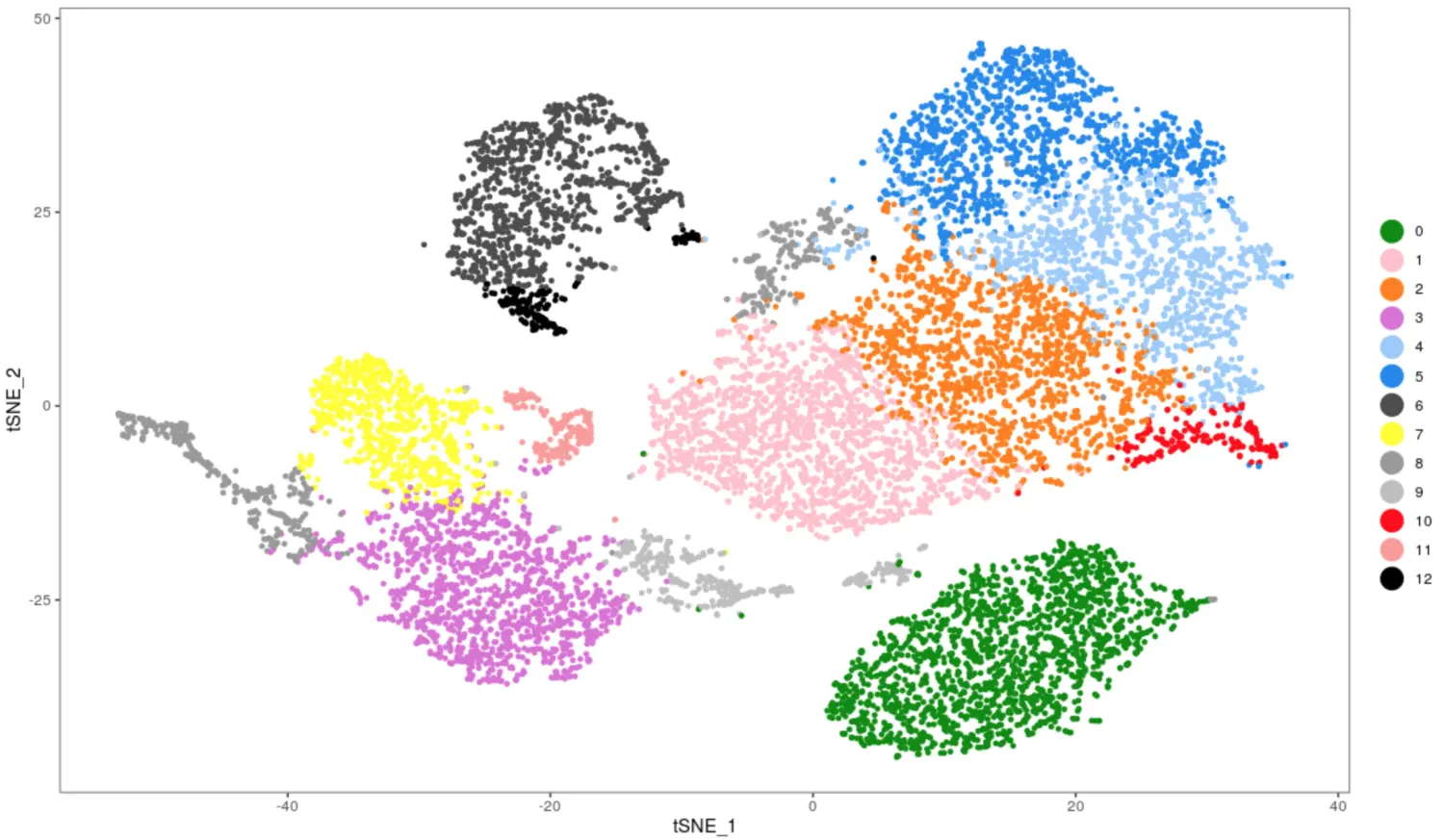
探究一下Seurat2和3的分群结果
起初我认为这两个版本的差异蛮大的,因为看tsne图明显感觉V2更好一些,导致我得出了错误的结论,认为两个版本的包处理结果千差万别。实际上,站在作者的角度思考一下,他也不会允许自己的“孩子”在成长过程中出现太大的偏差
学到了一点:图片不能说明问题 ,需要用数据来表现它们之间的分群结果到底差异大不大
下面👇就用数据来证明:
V2的Seurat得到了13群,V3利用resolution = 0.9得到了13群,而使用和V2一样的resolution = 1 会得到14群
主要使用table看一下比较结果
如果直接使用table(PBMC@meta.data$res.1),那么就会出现以下情况:
> table(PBMC@meta.data$res.1)
0 1 10 11 12 2 3 4 5 6 7 8 9
1877 1859 219 212 191 1569 1414 1322 1321 1123 772 595 400
# 发现虽然是13群,但10、11、12出现在了1以后,很明显这是由于字符串ascii排序的原因,在linux中常见
> class(PBMC@meta.data$res.1)
[1] "character"
# 使用数值型就会和平常一样
> table(as.numeric(PBMC@meta.data$res.1))
0 1 2 3 4 5 6 7 8 9 10 11 12
1877 1859 1569 1414 1322 1321 1123 772 595 400 219 212 191
横着的0-12是V2得到的PBMC,竖着的0-12是V3得到的PBMC_V3。看到V2的第1群在V3中分成了第4和第2群【在图中显示就是原文图中粉色群 = V3得到的橙色和浅蓝色(如下图)】;V2的第6群在V3中分成了第6和9群
> table(PBMC_V3$RNA_snn_res.0.9,as.numeric(PBMC@meta.data$res.1))
0 1 2 3 4 5 6 7 8 9 10 11 12
0 1873 1 0 0 0 0 0 0 3 7 0 0 0
1 0 0 224 0 1094 277 0 0 10 1 3 0 1
2 0 420 1101 0 37 2 0 0 4 2 40 0 0
3 0 0 0 1379 0 0 0 82 60 17 0 5 0
4 0 1418 53 0 0 0 0 0 0 7 15 0 0
5 0 2 47 0 93 1029 0 0 6 0 1 1 0
6 0 0 1 0 1 0 887 0 0 0 0 0 32
7 0 0 0 13 0 0 0 648 0 6 0 2 0
8 1 17 3 3 21 11 0 12 500 5 0 0 0
9 1 1 0 1 0 0 236 0 2 0 0 0 158
10 2 0 1 11 1 0 0 12 1 354 2 1 0
11 0 0 139 0 75 2 0 0 9 1 158 0 0
12 0 0 0 7 0 0 0 18 0 0 0 203 0
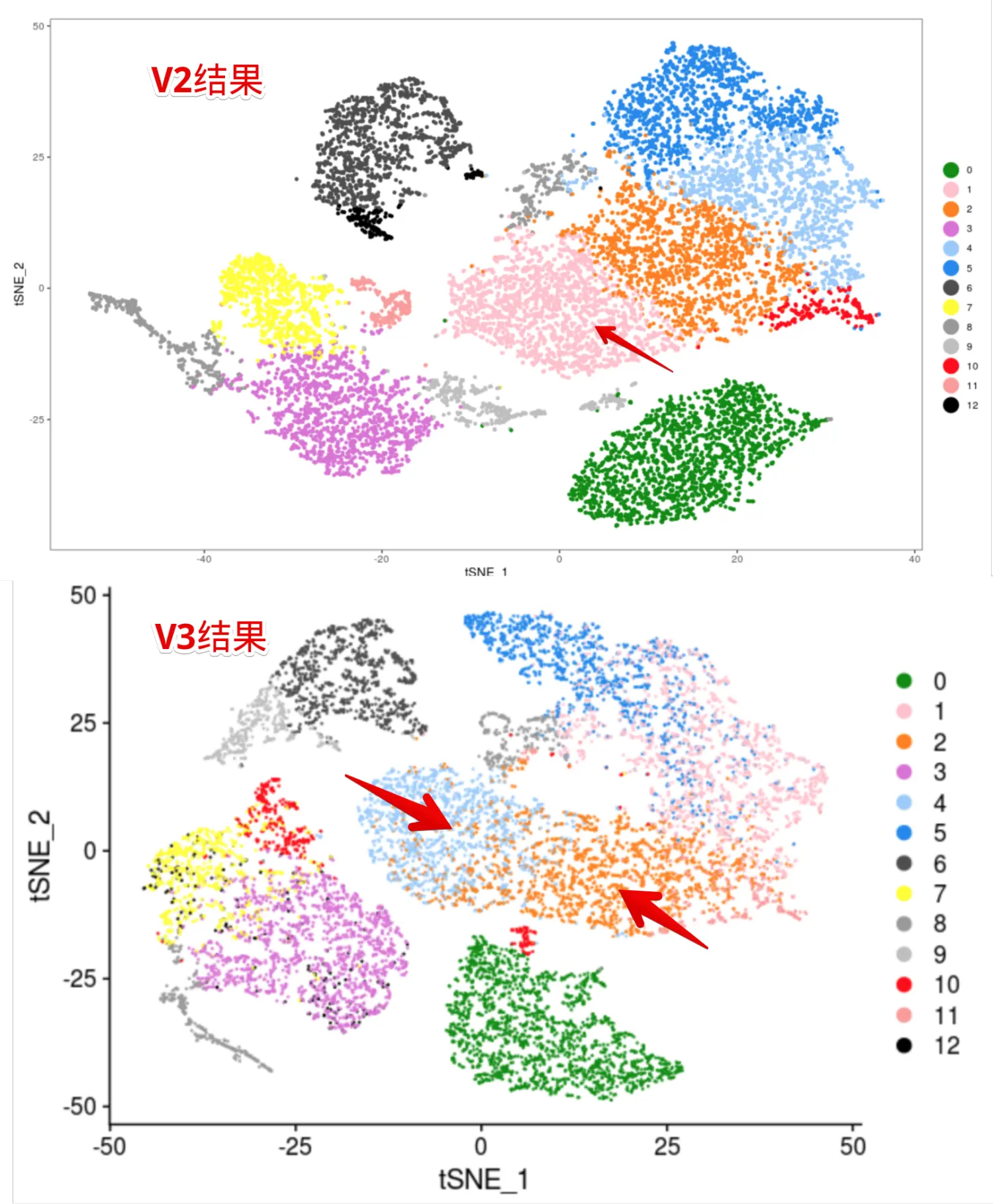
但是,这个结果说明了**:V2、V3处理同一组数据,结果其实相差并不大**。并不是像我们想象的,图片差距大,结果差距就大。真实情况可能是本来的某一群在另一种处理中被分得离散一些。但是tSNE本来就是这样,图中的距离并不代表真实的差异,它的运行次数会直接导致最后的图片形态不同
关于tsne这个流行的算法,有必要了解一下:
tsne的作者Laurens强调,可以通过
t-SNE的可视化图提出一些假设,但是不要用t-SNE来得出一些结论,想要验证你的想法,最好用一些其他的办法。t-SNE中集群之间的距离并不表示相似度 ,同一个数据上运行
t-SNE算法多次,很有可能得到多个不同“形态”的集群。但话说回来,真正有差异的群体之间,不管怎么变换形态,它们还是有差别关于perplexity的使用:(默认值是30) 如果忽视了perplexity带来的影响,有的时候遇到
t-SNE可视化效果不好时,对于问题无从下手。perplexity表示了近邻的数量,例如设perplexity为2,那么就很有可能得到很多两个一对的小集群。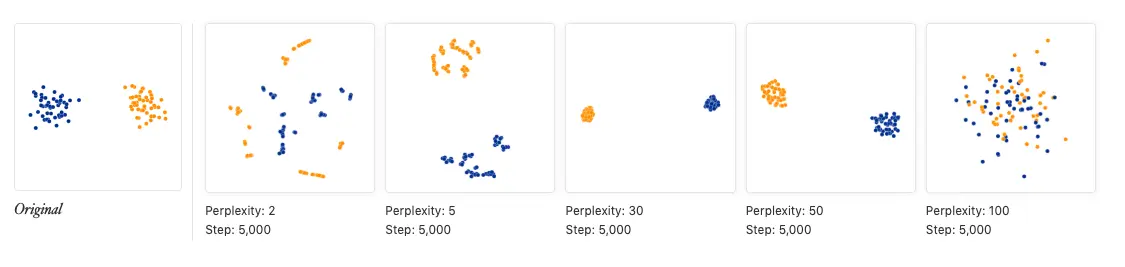
有的时候会出现同一集群被分为两半的情况,但群间的距离并不能说明什么,解决这个问题,只需要跑多次找出效果最好的就可以了
引用自: https://bindog.github.io/blog/2018/07/31/t-sne-tips/ 很好的tsne可视化:https://distill.pub/2016/misread-tsne/c
结尾
Seurat两个版本的结果的确存在不同,但不至于差异太大。它们的tSNE聚类结果“看似差异大”,其实是我们误认为tSNE图中的点之间距离代表了相似性。就像上面V3图中的两群分开的红点,它们其实还是一群,只不过此时此刻的映射坐标是这样的,而V2的映射坐标正好把它们映射到了相近的位置而已。也许多运行几次,使用不同的
perplexity参数,V3的结果又会发生变化数据胜于图片,使用
table比较是相对于肉眼观察图片更有效的方式