scRNA-10X免疫治疗学习笔记-2-配置Seurat的R语言环境
刘小泽写于19.10.14 笔记目的:根据生信技能树的单细胞转录组课程探索10X Genomics技术相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 第二单元第6讲:配置Seurat的R语言环境
下游分析前言
下游分析一般是研究的重点,之前10X上游得到的结果中,对我们最有用的是三个文件和一个报告
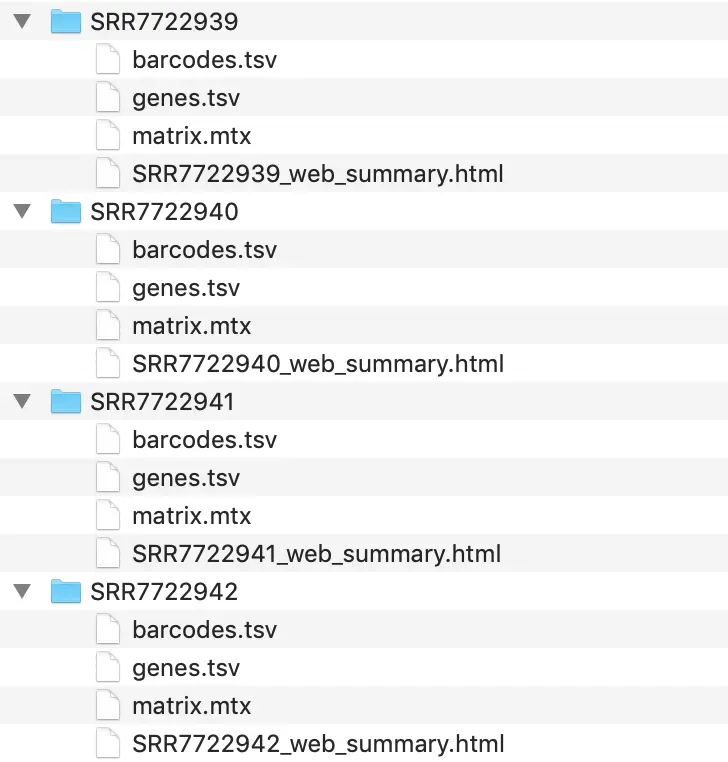
这篇文章的作者其实已经把表达矩阵上传到了 GSE117988:
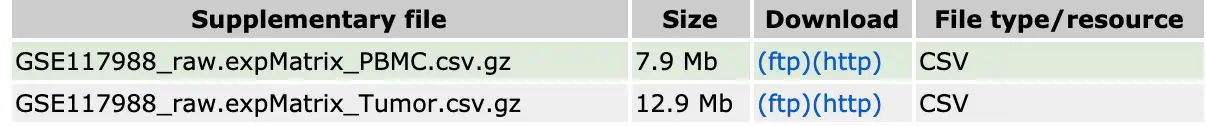
但为了演示如何组合多个样本数据,还是使用了自己之前用cellranger得到的4个count结果
下面会组合四个PBMC数据
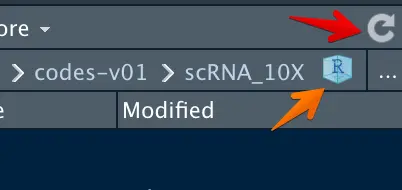
打开Rstudio后注意一个小技巧
红色箭头指的是刷新,当有新文件生成时,记得刷新一下,否则文件名有时显示不出来;
橙色箭头指的是回到最开始定位的路径
首先做一件事——Seurat降级
原因就是这篇文章使用的是Seurat V2,想用V3也可以做,只不过想更好地重现文章代码
两步走:删除原文件(直接
remove.packages删掉原来包的文件夹)+ 源代码安装
remove.packages('Seurat')
pkgs = c( 'mixtools', 'lars', 'dtw', 'doSNOW', 'hdf5r' )
BiocManager::install(pkgs,ask = F,update = F)
# 以后只需要修改这个版本号即可
packageurl <- "https://cran.r-project.org/src/contrib/Archive/Seurat/Seurat_2.3.4.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
# 最后检查一下包的版本
library('Seurat')
packageVersion('Seurat')
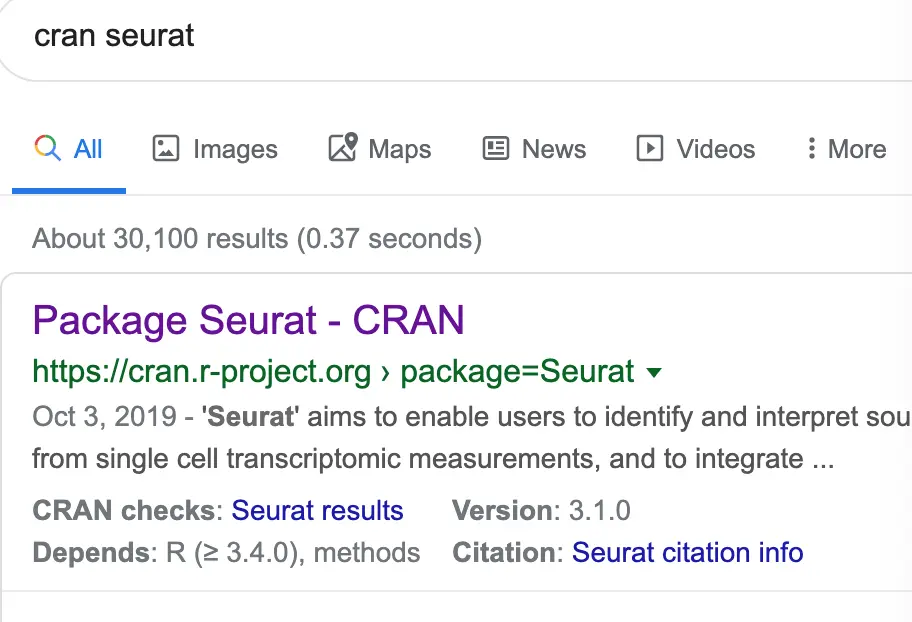
有个问题,怎么自己去寻找要安装包的路径呢?
step1:首先浏览器搜索:cran + 包的名称 ,比如我要搜索seurat,就会看到:
step2:进来后,看到有一个Old sources:选项,点开链接

step3:自由安装想要的版本(右键复制链接赋给上面的packageurl)
接下来分别读取
library(Seurat)
sce.10x <- Read10X(data.dir = '~/four-PBMC-mtx/SRR7722939/')
sce1 <- CreateSeuratObject(raw.data = sce.10x,
min.cells = 60,
min.genes = 200,
project = "SRR7722939")
sce.10x <- Read10X(data.dir = '~/four-PBMC-mtx/SRR7722940/')
sce2 <- CreateSeuratObject(raw.data = sce.10x,
min.cells = 60,
min.genes = 200,
project = "SRR7722940")
sce.10x <- Read10X(data.dir = '~/four-PBMC-mtx/SRR7722941/')
sce3 <- CreateSeuratObject(raw.data = sce.10x,
min.cells = 60,
min.genes = 200,
project = "SRR7722941")
sce.10x <- Read10X(data.dir = '~/four-PBMC-mtx/SRR7722942/')
sce4 <- CreateSeuratObject(raw.data = sce.10x,
min.cells = 60,
min.genes = 200,
project = "SRR7722942")
sce1;sce2;sce3;sce4
## An object of class seurat in project SRR7722939
## 6163 genes across 2047 samples.
## An object of class seurat in project SRR7722940
## 4267 genes across 1073 samples.
## An object of class seurat in project SRR7722941
## 5480 genes across 4311 samples.
## An object of class seurat in project SRR7722942
## 6427 genes across 4025 samples.
或者采用批量处理的方式:
folders=list.files('../../cellranger/four-PBMC-mtx/',pattern = '^SRR')
> folders
[1] "SRR7722939" "SRR7722940" "SRR7722941" "SRR7722942"
library(Seurat)
sceList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder , min.cells = 60,
min.features = 200)
})
读取后进行区分
四个数据分别对应了四个时间点:
- 治疗前(Pre): GSM3330561
- 治疗后早期day +27(Early): GSM3330562
- 治疗后反应期day+37(Resp): GSM3330563
- 治疗后复发+614 (AR): GSM3330564
区分对象名称
根据这个信息,修改一下读入数据的编号(原来记录的是SRR信息,但这个信息不足以区分四个数据)
sce1@meta.data$group <- "PBMC_Pre"
sce2@meta.data$group <- "PBMC_EarlyD27"
sce3@meta.data$group <- "PBMC_RespD376"
sce4@meta.data$group <- "PBMC_ARD614"
区分细胞名
然后看看它们的列名,记录的就是细胞barcode信息,区分不同细胞,因此前面看到的sce1有2047个细胞就是说明sce1有2047个有效barcode【注意这里是“有效”,对应之前创建对象时设定的阈值:一个细胞中要有多少基因表达min.genes和一个基因要在多少细胞中表达min.cells】
# 以sce1对象为例
head(colnames(sce1@data))
## [1] "AAACCTGAGCGAAGGG" "AAACCTGAGGTCATCT" "AAACCTGAGTCCTCCT"
## [4] "AAACCTGCACCAGCAC" "AAACCTGGTAACGTTC" "AAACCTGGTAAGGATT"
将四个对象对应的名称添加到细胞名中:
colnames(sce1@data) <- paste0("PBMC_Pre.",colnames(sce1@data))
head(colnames(sce1@data))
## [1] "PBMC_Pre.AAACCTGAGCGAAGGG" "PBMC_Pre.AAACCTGAGGTCATCT"
## [3] "PBMC_Pre.AAACCTGAGTCCTCCT" "PBMC_Pre.AAACCTGCACCAGCAC"
## [5] "PBMC_Pre.AAACCTGGTAACGTTC" "PBMC_Pre.AAACCTGGTAAGGATT"
colnames(sce2@data) <- paste0("PBMC_EarlyD27.",colnames(sce2@data))
colnames(sce3@data) <- paste0("PBMC_RespD376.",colnames(sce3@data))
colnames(sce4@data) <- paste0("PBMC_ARD614.",colnames(sce4@data))
然后进行整合
归一化+标准化 => NormalizeData + ScaleData
sce1 <- NormalizeData(sce1)
sce1 <- ScaleData(sce1, display.progress = F)
sce2 <- NormalizeData(sce2)
sce2 <- ScaleData(sce2, display.progress = F)
sce3 <- NormalizeData(sce3)
sce3 <- ScaleData(sce3, display.progress = F)
sce4 <- NormalizeData(sce4)
sce4 <- ScaleData(sce4, display.progress = F)
找共有的高变异基因 => FindVariableGenes
做这一步的目的就是:利用共有的基因去进行后面的校正,如果一个基因这个数据集中有,而另一个没有,那么就很难根据这个基因对它们进行校正
sce1 <- FindVariableGenes(sce1, do.plot = F)
sce2 <- FindVariableGenes(sce2, do.plot = F)
sce3 <- FindVariableGenes(sce3, do.plot = F)
sce4 <- FindVariableGenes(sce4, do.plot = F)
# 前1000个HVGs结果记录下来
g.1 <- head(rownames(sce1@hvg.info), 1000)
g.2 <- head(rownames(sce2@hvg.info), 1000)
g.3 <- head(rownames(sce3@hvg.info), 1000)
g.4 <- head(rownames(sce4@hvg.info), 1000)
# 再取交集
genes.use <- unique(c(g.1, g.2,g.3, g.4))
genes.use <- intersect(genes.use, rownames(sce1@scale.data))
genes.use <- intersect(genes.use, rownames(sce2@scale.data))
genes.use <- intersect(genes.use, rownames(sce3@scale.data))
genes.use <- intersect(genes.use, rownames(sce4@scale.data))
head(genes.use)
## [1] "hg38_S100A9" "hg38_PPBP" "hg38_S100A8" "hg38_MALAT1"
## [5] "hg38_PF4" "hg38_HIST1H2AC"
# 最后得到1450个共有高变异基因
length(genes.use)
## [1] 1450
10X 数据集的合并 => RunMultiCCA
这一步耗时很长
sce.comb <- RunMultiCCA(list(sce1,sce2,sce3,sce4),
add.cell.ids=c("PBMC_Pre.","PBMC_EarlyD27.","PBMC_RespD376.","PBMC_ARD614."),
genes.use = genes.use,
num.cc = 30)
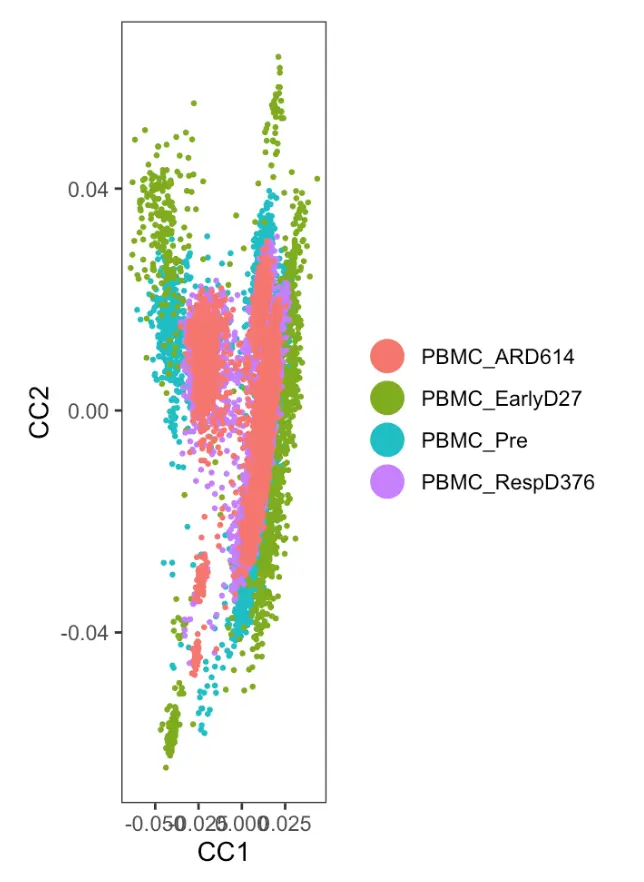
DimPlot(object = sce.comb,
reduction.use = "cca", group.by = "group",
pt.size = 0.5, do.return = TRUE)
看到CCA并没有有效去除批次效应,按说四个时间点的细胞应该重叠在一起,表示某群细胞在四个时间点都存在。不过这里绿色和红色的细胞分的很开
再试一下另一种办法=>merge + SCTransform
# 以下代码基于Seurat 3.1
sceList[[1]]@meta.data$orig.ident <- "PBMC_Pre"
sceList[[2]]@meta.data$orig.ident <- "PBMC_EarlyD27"
sceList[[3]]@meta.data$orig.ident <- "PBMC_RespD376"
sceList[[4]]@meta.data$orig.ident <- "PBMC_ARD614"
sce.big <- merge(sceList[[1]],
y = c(sceList[[2]],sceList[[3]],sceList[[4]]),
add.cell.ids = c("PBMC_Pre.","PBMC_EarlyD27.","PBMC_RespD376.","PBMC_ARD614."),
project = "p1-PBMC")
> table(sce.big$orig.ident)
PBMC_Pre SRR7722940 SRR7722941 SRR7722942
2047 1074 4311 4028
sce.big <- SCTransform(sce.big, verbose = FALSE)
sce.big <- RunPCA(sce.big, verbose = FALSE)
sce.big <- RunTSNE(sce.big, verbose = FALSE)
DimPlot(object = sce.big,
reduction = "tsne",group.by = 'orig.ident')
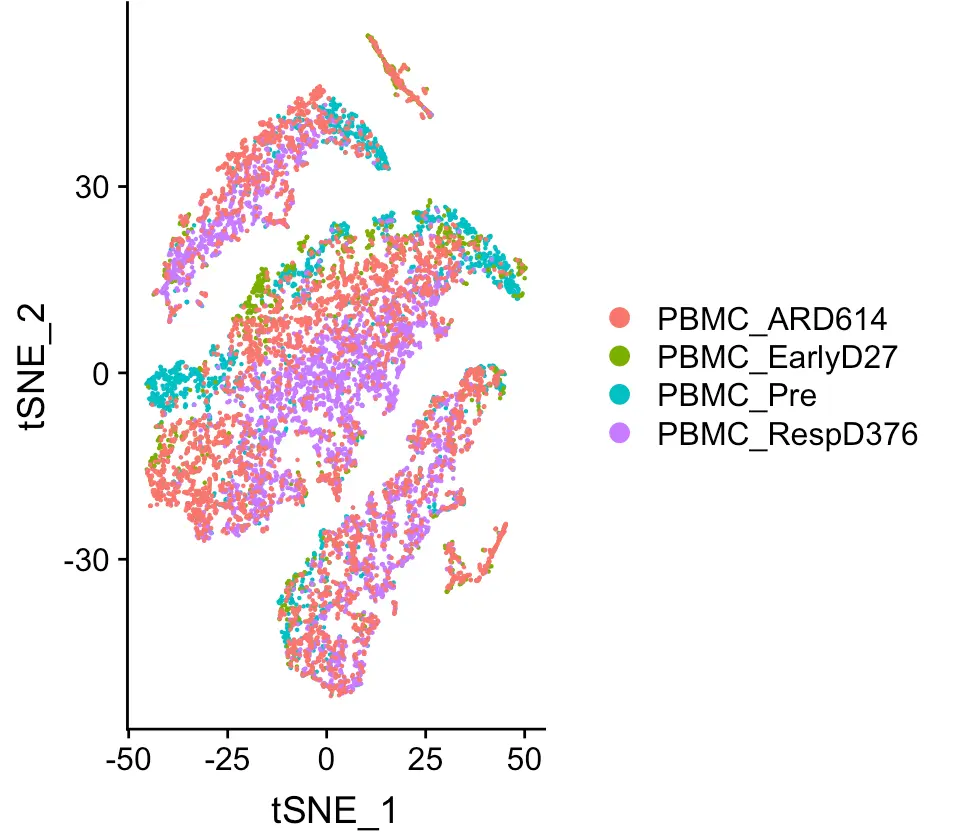
之后鉴定亚群
sce.big <- FindNeighbors(sce.big, dims = 1:20)
sce.big <- FindClusters(sce.big, resolution = 0.2)
table(sce.big@meta.data$SCT_snn_res.0.2)
DimPlot(object = sce.big, reduction = "tsne",
group.by = 'SCT_snn_res.0.2')

> table(sce.big$SCT_snn_res.0.2,sce.big$orig.ident)
PBMC_ARD614 PBMC_EarlyD27 PBMC_Pre PBMC_RespD376
0 1175 347 22 1555
1 987 152 137 1103
2 862 35 24 488
3 396 43 14 471
4 0 2 862 1
5 294 0 160 330
6 26 272 6 261
7 0 1 361 1
8 1 43 228 2
9 112 130 1 15
10 0 0 205 5
11 134 29 4 29
12 41 20 23 50