scRNA-小鼠发育学习笔记-6-不同谱系的差异基因分类注释
刘小泽写于19.10.24 笔记目的:根据生信技能树的单细胞转录组课程探索Smartseq2技术及发育相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 这次会介绍如何对不同谱系的差异基因分类注释。对应视频第三单元14讲
前言
将对应文章这张图:
1 Monocle找不同谱系之间的高变化基因
加载数据
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
source("../analysis_functions.R")
# RPKM矩阵进行可视化,count矩阵进行差异分析
load('../female_rpkm.Rdata')
load('../female_count.Rdata')
# 谱系推断结果,这里选择之前的Slingshot结果
load('../step4-psudotime/female-psudotime-percent.Rdata')
# 6个发育时期获取
head(colnames(female_count))
female_stages <- sapply(strsplit(colnames(female_count), "_"), `[`, 1)
names(female_stages) <- colnames(female_count)
table(female_stages)
# 4个cluster获取
cluster <- read.csv('../step1-female-RPKM-tSNE/female_clustering.csv')
female_clustering=cluster[,2];names(female_clustering)=cluster[,1]
table(female_clustering)
首先找第一个谱系中变化更剧烈的
下面这个get_var_genes_pseudotime函数是作者包装好的(https://github.com/IStevant/XX-XY-mouse-gonad-scRNA-seq/blob/master/scripts/XX_analysis_dm.R),很长但不难理解。只需要自己进入作者的代码,将其中的变量替换成自己现有的变量,一步步操作理解即可。
female_lineage1_sig_gene_pseudoT <- get_var_genes_pseudotime(
females,
female_count,
female_pseudotime,
lineageNb=1,
female_clustering
)
> dim(female_lineage1_sig_gene_pseudoT)
[1] 12612 6
# 从中找到差异显著的基因,根据qval<0.05过滤
# 结果从12612个基因里面挑选出2861个差异显著的基因
female_lineage1_sig_gene_pseudoT <- female_lineage1_sig_gene_pseudoT[female_lineage1_sig_gene_pseudoT$qval<0.05,]
> dim(female_lineage1_sig_gene_pseudoT)
[1] 2861 6
同样对第二个谱系
female_lineage2_sig_gene_pseudoT <- get_var_genes_pseudotime(
females,
female_count,
female_pseudotime,
lineageNb=2,
female_clustering
)
female_lineage2_sig_gene_pseudoT <- female_lineage2_sig_gene_pseudoT[female_lineage2_sig_gene_pseudoT$qval<0.05,]
# 从11937个基因里面挑选出2182个差异显著的基因
> dim(female_lineage2_sig_gene_pseudoT)
[1] 2182 6
save(female_lineage1_sig_gene_pseudoT,
female_lineage2_sig_gene_pseudoT,
file = 'lineage_sig_gene.Rdata')
2 将不同谱系中的高变化基因进行分类
找到了变化显著的基因,就相当于缩小了操作对象,下面聚类的操作就会得到这些基因并基于它们进行后续分析
2.1 取两个谱系全部的HVGs,并进行去重复
首先各自提取两个谱系中差异显著的基因
female_lineage1_clustering <- female_lineage1_sig_gene_pseudoT[female_lineage1_sig_gene_pseudoT$qval<0.05,]
female_lineage2_clustering <- female_lineage2_sig_gene_pseudoT[female_lineage2_sig_gene_pseudoT$qval<0.05,]
然后得到之前两个谱系的全部高变化基因,并使用unique()函数进行去重复
理解一下这个函数。对于重复值操作,常见的有unique和duplicated,前者直接返回去重复后的结果;后者返回逻辑值,判断是否为重复值
> unique(c(1,2,1,2,3))
[1] 1 2 3
> duplicated(c(1,2,1,2,3))
[1] FALSE FALSE TRUE TRUE FALSE
那么就可以得到全部高变化基因
gene_list <- unique(rownames(female_lineage1_clustering),
rownames(female_lineage2_clustering))
> length(gene_list)
[1] 2861
这样就可以提取无重复HVGs的RPKM小表达矩阵
de_matrix <- log(females[rownames(females) %in% gene_list,]+1)
> dim(females)
[1] 21083 563
> dim(de_matrix)
[1] 2861 563
最后得到2861个高变化基因的小表达矩阵
2.2 分别得到两个谱系的细胞排序后的表达矩阵
**思路:**从开始的左边👈那张图可以看到,两个谱系的细胞都是从中间0开始向两侧(100)延伸,那么这里也需要按照之前做好的谱系百分比对细胞进行一个升序排序,然后再按照这个顺序提取每个谱系的表达矩阵
## 对第一个谱系来说
# 得到第一个谱系的细胞百分比
L1_lineage <- female_pseudotime[!is.na(female_pseudotime[,1]),1]
# 对百分比进行升序排序
L1_ordered_lineage <- L1_lineage[order(L1_lineage,
decreasing = FALSE)]
# 根据第一个谱系的排序后的细胞名称,得到属于它的表达矩阵
L1_cells <- de_matrix[,names(L1_ordered_lineage)]
## 同理得到L2
if(T){
## 第二个谱系
# 得到第二个谱系的细胞百分比
L2_lineage <- female_pseudotime[!is.na(female_pseudotime[,2]),2]
# 对百分比进行升序排序(细胞数量从少到多)
L2_ordered_lineage <- L2_lineage[order(L2_lineage,
decreasing = FALSE)]
# 根据第二个谱系的细胞名称,得到属于它的表达矩阵
L2_cells <- de_matrix[,names(L2_ordered_lineage)]
}
2.3 按照表达矩阵的细胞顺序,对谱系和分群的细胞名重新排序
把这两个谱系的排序好的细胞名称提取出来
## 提取细胞名
L1_lineage_cells <- names(L1_ordered_lineage)
length(L1_lineage_cells)
# 423
L2_lineage_cells <- names(L2_ordered_lineage)
length(L2_lineage_cells)
# 294
看到L1_lineage有423个,L2_lineage有294个,而总共563个细胞。
那么我们想知道,哪些细胞是两个谱系分化之前共有的,哪些是特有的
也就是找交集和补集
comp_list <- comparelists(L1_lineage_cells, L2_lineage_cells)
common_cells <- comp_list$intersect
L1_spe_cells <- L1_lineage_cells[!L1_lineage_cells %in% comp_list$intersect]
L2_spe_cells <- L2_lineage_cells[!L2_lineage_cells %in% comp_list$intersect]
length(common_cells);length(L1_spe_cells);length(L2_spe_cells)
# 共有的是154个,L1特有269个,L2特有140个
把L1特有和共有的标记成L1_cellLin
L1_cellLin <- c(
rep_along("common cells", common_cells),
rep_along("L1 cells", L1_spe_cells)
)
names(L1_cellLin) <- c(common_cells, L1_spe_cells)
然后按照之前L1的小表达矩阵L1_cells的列名进行重新排序
L1_cellLin <- L1_cellLin[match(colnames(L1_cells),names(L1_cellLin) )]
对L2也进行同样的操作,得到L2_cellLin
if(T){
L2_cellLin <- c(
rep_along("common cells", common_cells),
rep_along("L2 cells", L2_spe_cells)
)
names(L2_cellLin) <- c(common_cells, L2_spe_cells)
# 将L1_cellLin按照之前得到的L1表达矩阵列名重新排序
L2_cellLin <- L2_cellLin[match(colnames(L2_cells),names(L2_cellLin) )]
}
接着按照之前分群的结果对小表达矩阵的列名重新排序,同时也对上面的谱系顺序再次排序。
看到这么多次,反反复复的排序,目的就一个:把小表达矩阵、分群、细胞谱系的细胞信息做到对应
# 将第一个谱系的表达矩阵细胞名与分群、谱系信息联系起来
cellType_L1 <- female_clustering[colnames(L1_cells)]
colnames(L1_cells) <- paste(colnames(L1_cells), "L1", sep="_")
names(L1_cellLin) <- colnames(L1_cells)
# 同理对L2
cellType_L2 <- female_clustering[colnames(L2_cells)]
colnames(L2_cells) <- paste(colnames(L2_cells), "L2", sep="_")
names(L2_cellLin) <- colnames(L2_cells)
然后合并排序后的谱系和分群信息
cellLin <- c(
L2_cellLin,
L1_cellLin
)
cellType <- c(
cellType_L2,
cellType_L1
)
2.4 开始对基因进行分类
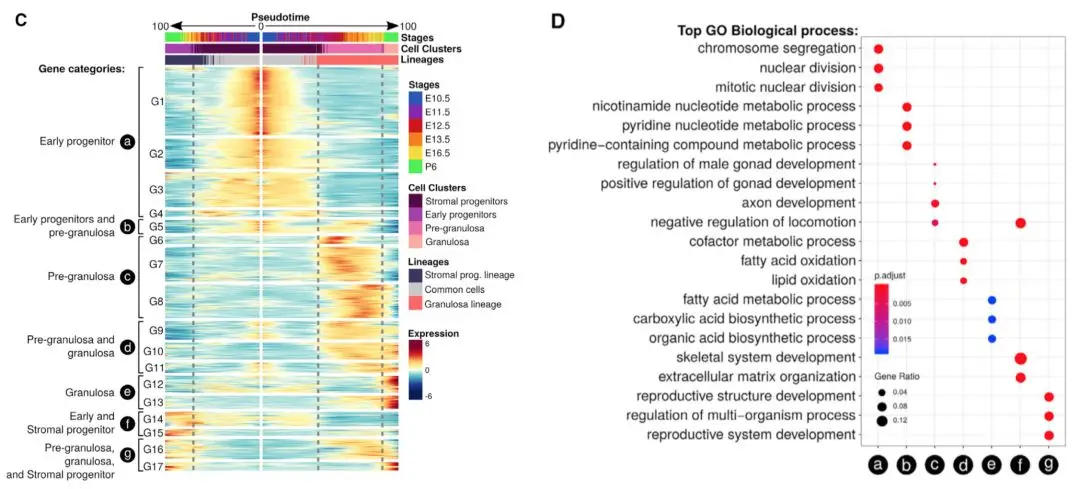
从图中可以看到,将基因 分成了17组
# 表达量局部加权回归散点平滑法(locally weighted scatterplot smoothing,LOESS)
L2_cells_smooth <- smooth_gene_exp(
L2_cells,
L2_ordered_lineage,
span=0.4
)
L1_cells_smooth <- smooth_gene_exp(
L1_cells,
L1_ordered_lineage,
span=0.4
)
# 合并局部加权回归表达矩阵
data_heatmap <- data.frame(
L2_cells_smooth,
L1_cells_smooth
)
# 利用pheatmap函数进行层次聚类,只是为了调用它的算法而已,不是真的作图
set.seed(123)
gene_clustering <- pheatmap::pheatmap(
data_heatmap,
scale="row",
clustering_method="ward.D",
silent=TRUE
)
# 挑出17个基因cluster
clusters <- cutree(gene_clustering$tree_row, k = 17)
clustering <- data.frame(clusters)
clustering[,1] <- as.character(clustering[,1])
colnames(clustering) <- "Gene_Clusters"
write.csv(clustering,
file="step5.2-gene_clustering_kmeans_k17_scaled.csv")
save(clusters,clustering,file = 'step5.2-gene_clustering.Rdata')
save(data_heatmap,cellLin,cellType,cellType_L2,
file = 'step5.2-for_heatmap.Rdata')
3 分类后,添加颜色信息进行热图绘制
3.1 首先还是添加之前做好的数据
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
source("../analysis_functions.R")
load('step5.2-for_heatmap.Rdata')
load('step5.2-gene_clustering.Rdata')
# 3个谱系(2个特有+1个共有)
> table(cellLin)
cellLin
L1 cells L2 cells common cells
269 140 308
# 4种细胞类型
> table(cellType)
cellType
C1 C2 C3 C4
394 90 190 43
# 17个基因分类结果
> table(clustering[,1])
1 10 11 12 13 14 15 16 17 2 3 4 5 6 7 8 9
295 257 117 69 122 128 209 162 150 319 61 167 83 136 229 129 228
3.2 热图准备之–配置基因分组颜色
gene_cluster_palette <- c(
'#a6cee3',
'#1f78b4',
'#b2df8a',
'#33a02c',
'#fb9a99',
'#e31a1c',
'#fdbf6f',
'#ff7f00',
'#cab2d6',
'#6a3d9a',
'#ffff99',
'#b15928',
'#49beaa',
'#611c35',
'#2708a0',
'#fccde5',
'#bc80bd'
)
gene_cluster_colors <- gene_cluster_palette[1:max(clusters)]
names(gene_cluster_colors) <- 1:max(clusters)
3.3 热图准备之–配置行、列注释信息
行注释:每个基因属于哪个组
annotation_row <- data.frame(clustering=clustering)
列注释:三种信息cell lineage, cell cluster type, cell stage
annotation_col <- data.frame(
cellLineages=cellLin,
cellType=cellType,
Stages=sapply(strsplit(colnames(data_heatmap), "_"), `[`, 1)
)
rownames(annotation_col) <- colnames(data_heatmap)
3个cell lineages颜色
cellLinCol <- c(
"#3b3561",
"#c8c8c8",
"#ff6663"
)
names(cellLinCol) <- unique(cellLin)
4个cell clusters颜色
cellTypeCol <- c(
C2="#a53bad",
C1="#560047",
C3="#eb6bac",
C4="#ffa8a0"
)
names(cellTypeCol) <- unique(cellType)
3.4 热图准备之–把三种注释信息颜色放在一起
3个谱系(2个特有+1个共有)+4种细胞类型 +17个基因分类结果
annotation_colors <- list(
cellType=cellTypeCol,
cellLineages=cellLinCol,
clustering=gene_cluster_colors,
Stages=c(
E10.5="#2754b5",
E11.5="#8a00b0",
E12.5="#d20e0f",
E13.5="#f77f05",
E16.5="#f9db21",
P6="#43f14b"
)
)
# 调画板
cold <- colorRampPalette(c('#f7fcf0','#41b6c4','#253494','#081d58','#081d58'))
warm <- colorRampPalette(c('#ffffb2','#fecc5c','#e31a1c','#800026','#800026'))
mypalette <- c(rev(cold(21)), warm(20))
breaksList = seq(-2.2, 2.5, by = 0.2)
3.5 绘制热图
library(pheatmap)
tiff(file="step5.3-A-female_heatmap_DE_genes_k_17_pval_005.tiff",
res = 300, height = 21, width = 18, units = 'cm')
gene_clustering <- pheatmap(
data_heatmap,
scale="row",
gaps_col=length(cellType_L2),
show_colnames=FALSE,
show_rownames=FALSE,
cluster_cols=FALSE,
clustering_method="ward.D",
annotation_row=annotation_row,
annotation_col=annotation_col,
annotation_colors=annotation_colors,
cutree_rows=17,
annotation_names_row=FALSE,
color=mypalette
)
dev.off()

4 功能分析
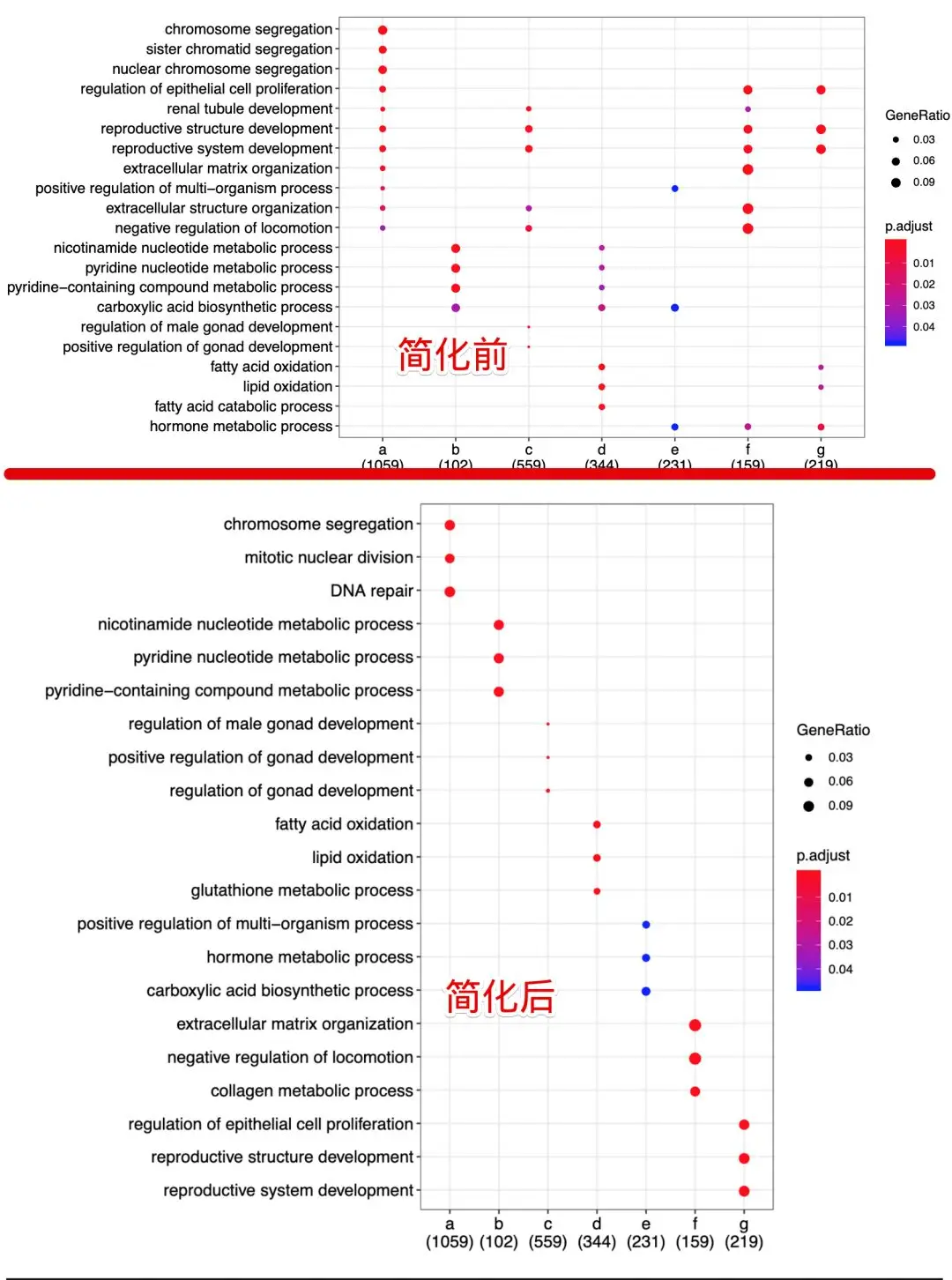
上一步将基因分成了G1-G17组,然后作者根据相似的表达模式又进行整合,再看原文的那张图,将G1-G4规定为a(从热图中能看到它们都在早期表达,在晚期不表达),类似地分成了a-g7组。分组的原因一个是:原来的17组进行注释太繁琐;另一个是:原来的17组中有的组细胞数量太少,注释结果也不好解释。正好借助热图,观察到有的组很像,那么就干脆将它们放在一起进行注释。新的分组也是有意义的,文章中也花了大篇幅介绍这些整合是根据什么:

下载作者做好的分组数据
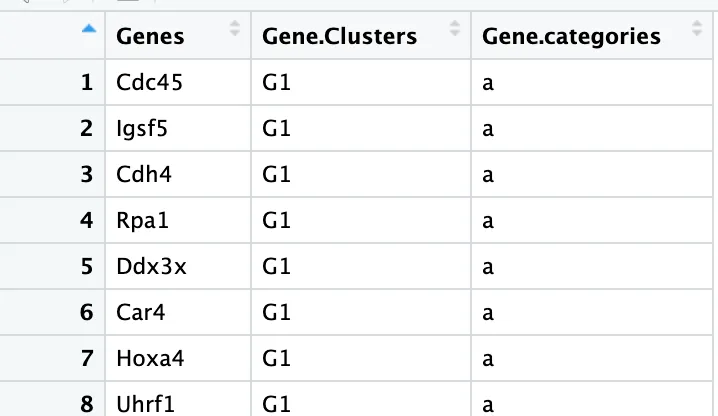
dyn_genes <- read.csv(file="../female_lineages_DE_gene_pseudotime_clustered_annotated.csv")
gene_names <- dyn_genes$Genes
基因ID转换
entrez_genes <- bitr(gene_names, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Mm.eg.db")
提取有对应Entrez ID的变化基因
gene_clusters <- dyn_genes[dyn_genes$Genes %in% entrez_genes$SYMBOL,,drop=FALSE]
# drop=FALSE确保返回数据框
进行富集分析
de_gene_clusters <- data.frame(
ENTREZID=entrez_genes[!duplicated(entrez_genes$SYMBOL),"ENTREZID"],
Gene_Clusters=gene_clusters$Gene.categories
)
formula_res <- compareCluster(
ENTREZID~Gene_Clusters,
data=de_gene_clusters,
fun="enrichGO",
OrgDb="org.Mm.eg.db",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05
)
因为有7个分组,所以富集分析也是一个组一个组地去做,但是这里可以直接提供数据框格式,然后函数本身再对数据框进行拆分成列表的操作,只是方便了使用,背后的逻辑没有变
它在背后做了:
split(de_gene_clusters,de_gene_clusters$Gene_Clusters)
简化GO富集分析结果
GO数据库是有向无环图,存在父-子关系,因此常规的注释可能会注释到很多同属一个根的结果,会有些冗余。可以选择使用simplify函数进行简化
lineage1_ego <- simplify(
formula_res,
cutoff=0.5,
by="p.adjust",
select_fun=min
)
结果可以对比一下
dotplot(formula_res, showCategory=3)+ theme(aspect.ratio=0.8)
dotplot(lineage1_ego, showCategory=3)+ theme(aspect.ratio=2)