scRNA-小鼠发育学习笔记-4-差异分析及功能注释
刘小泽写于19.10.24 笔记目的:根据生信技能树的单细胞转录组课程探索Smartseq2技术及发育相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 这次会介绍如何进行差异分析及功能注释,以及比较不同分组。对应视频第三单元9-11讲
前言
前面得到的 6个发育时期和4个分群,而且还 可视化了一些marker基因,那么现在就要对这4群细胞进行差异分析
将对应文章这张图:
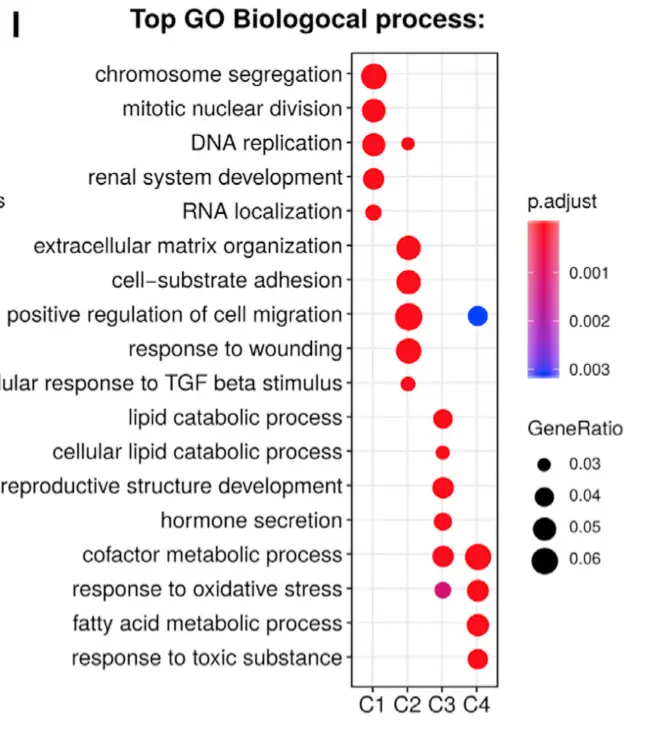
1 使用monocle V2进行差异分析
1.1 准备表达矩阵和分群信息
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
source("../analysis_functions.R")
# 差异分析一般都要求使用count矩阵
load('../female_count.Rdata')
# 6个发育时期获取
head(colnames(female_count))
female_stages <- sapply(strsplit(colnames(female_count), "_"), `[`, 1)
names(female_stages) <- colnames(female_count)
table(female_stages)
# 4个cluster获取
cluster <- read.csv('female_clustering.csv')
female_clustering=cluster[,2];names(female_clustering)=cluster[,1]
table(female_clustering)
1.2 利用monocle V2构建对象
需要三样东西:表达矩阵、细胞信息、基因信息
## 表达矩阵
dim(female_count)
## 细胞分群信息(包括6个stage和4个cluster)
table(female_stages)
table(female_clustering)
## 基因信息
gene_annotation <- as.data.frame(rownames(count_matrix))
开始构建
library(monocle)
# 直接使用作者包装的函数,代码更简洁
DE_female <- prepare_for_DE (
female_count,
female_clustering,
female_stages
)
可以看到,包装好的函数只需要提供表达矩阵和细胞信息,其实它背后做的事情比较多,就像下面👇的代码
# 1.表达矩阵
expr_matrix <- as.matrix(count_matrix)
# 2.细胞信息
sample_ann <- data.frame(cells=names(count_matrix),
stages=stage,
cellType=cluster)
rownames(sample_ann)<- names(count_matrix)
# 3.基因信息
gene_ann <- as.data.frame(rownames(count_matrix))
rownames(gene_ann)<- rownames(count_matrix)
colnames(gene_ann)<- "genes"
# 然后转换为AnnotatedDataFrame对象
pd <- new("AnnotatedDataFrame",
data=sample_ann)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
# 最后构建CDS对象
sc_cds <- newCellDataSet(
expr_matrix,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=0.5)
sc_cds <- detectGenes(sc_cds, min_expr = 5)
sc_cds <- sc_cds[fData(sc_cds)$num_cells_expressed > 10, ]
sc_cds <- estimateSizeFactors(sc_cds)
sc_cds <- estimateDispersions(sc_cds)
1.3 进行差异分析
其实也是包装了differentialGeneTest函数
start_time <- Sys.time()
female_DE_genes <- findDEgenes(
DE_female,
qvalue=0.05
)
end_time <- Sys.time()
end_time - start_time
# 1] "4435 significantly DE genes (FDR<0.05)."
# [1] "3268 significantly DE genes (FDR<0.01)."
# Time difference of 1.796054 mins
我们最后保留的也就是4435 significantly DE genes (FDR<0.05).
实际上,这个包装的函数就是下面这样,我们只需要提供创建好的monocle对象和q值即可:
function(HSMM=HSMM, qvalue=qvalue){
diff_test_res <- differentialGeneTest(
HSMM,
fullModelFormulaStr="~cellType",
cores = 3
)
sig_genes_0.05 <- subset(diff_test_res, qval < 0.05)
sig_genes_0.01 <- subset(diff_test_res, qval < 0.01)
# 帮我们统计了不同q值得到的差异基因数量
print(paste(nrow(sig_genes_0.05), " significantly DE genes (FDR<0.05).", sep=""))
print(paste(nrow(sig_genes_0.01), " significantly DE genes (FDR<0.01).", sep=""))
diff_test_res <- subset(diff_test_res, qval< qvalue)
return(diff_test_res)
}
最后得到这么一个差异基因数据框:
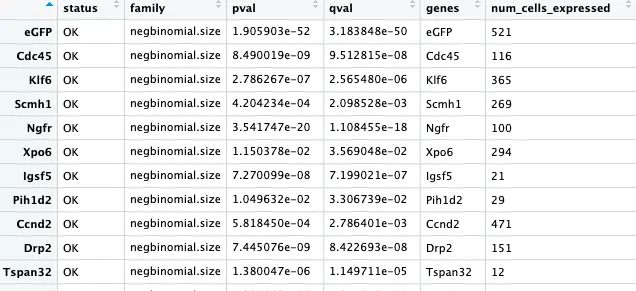
上面得到的4435个差异基因,我们要知道这些基因在哪个cluster:
作者的做法是:先得到了每个差异基因在不同cluster的平均表达量,然后找平均表达量最大的那个cluster,就认为这个基因属于这个cluster
# 有了差异基因以后,继续使用RPKM值
load('../female_rpkm.Rdata')
de_clusters <- get_up_reg_clusters(
females,
female_clustering,
female_DE_genes
)
2 使用ROTS进行差异分析
2.1 准备表达矩阵和分群信息
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
# 6个发育时期获取
head(colnames(female_count))
female_stages <- sapply(strsplit(colnames(female_count), "_"), `[`, 1)
names(female_stages) <- colnames(female_count)
table(female_stages)
# 4个cluster获取
cluster <- read.csv('female_clustering.csv')
female_clustering=cluster[,2];names(female_clustering)=cluster[,1]
table(female_clustering)
2.2 使用ROTS
下面会使用ROTS包(Reproducibility-optimized test statistic),对每个群和其他几个群的共同体进行比较
- ROTS可以使用RPKM值;它的速度可能会很慢
- 差异分析重点就在:表达矩阵和分组信息
配置最重要的分组信息
library(ROTS)
library(plyr)
# 使用RPKM矩阵
load('../female_rpkm.Rdata')
# 当前分组是这样
> table(female_clustering)
female_clustering
C1 C2 C3 C4
240 90 190 43
# 我们只需要用数字来表示,于是可以用substring(x, start, stop)获取
groups<-female_clustering
groups<-as.numeric(substring(as.character(groups),2,2))
> table(groups)
groups
1 2 3 4
240 90 190 43
# 如果要对第1群进行差异分析,那么就要把它和其他群比较(可将其他亚群定义为234)
groups[groups!=1]<-234
# 同理,对第2群进行差异分析
groups[groups!=2]<-134
然后配置表达矩阵
RPKM.full=females
ROTS_input<-RPKM.full[rowMeans(RPKM.full)>=1,]
ROTS_input<-as.matrix(log2(ROTS_input+1))
第1群
start_time <- Sys.time()
results_pop1 = ROTS(data = ROTS_input, groups = groups , B = 1000 , K = 500 , seed = 1234)
summary_pop1<-data.frame(summary(results_pop1, fdr=1))
end_time <- Sys.time()
(end_time - start_time)
# Time difference of 18.40462 mins
然后第2群、第3群、第4群
groups[groups!=2]<-134
start_time <- Sys.time()
results_pop2 = ROTS(data = ROTS_input, groups = groups , B = 1000 , K = 500 , seed = 1234)
summary_pop2<-data.frame(summary(results_pop2, fdr=1))
end_time <- Sys.time()
(end_time - start_time)
# 第3群
groups[groups!=3]<-124
start_time <- Sys.time()
results_pop3 = ROTS(data = ROTS_input, groups = groups , B = 1000 , K = 500 , seed = 1234)
summary_pop3<-data.frame(summary(results_pop3, fdr=1))
end_time <- Sys.time()
(end_time - start_time)
# 第4群
groups[groups!=4]<-123
start_time <- Sys.time()
results_pop4 = ROTS(data = ROTS_input, groups = groups , B = 1000 , K = 500 , seed = 1234)
summary_pop3<-data.frame(summary(results_pop4, fdr=1))
end_time <- Sys.time()
(end_time - start_time)
# 都得到以后,共同保存
save(summary_pop1,summary_pop2,summary_pop3,summary_pop4,
file = 'ROTS_summary.Rdata')
3 差异基因功能注释
差异基因得到的方法有很多,例如DESeq2、EdgeR、Wilcox、SCDE等等,真正有意义的差异基因,即使使用不同的方法,最后结果也不会相差太多
有了差异基因,然后就是去注释
3.1 先对monocle的结果进行注释
rm(list=ls())
options(stringsAsFactors = F)
library(clusterProfiler)
load(file = 'step3.1-DEG-monocle_summary.Rdata')
得到差异基因名
de_genes <- subset(de_clusters, qval<0.05)
> length(de_genes$genes)
[1] 4435
然后基因ID转换
entrez_genes <- bitr(de_genes$genes, fromType="SYMBOL",
toType="ENTREZID",
OrgDb="org.Mm.eg.db")
作者剔除掉一个基因名
entrez_genes <- entrez_genes[!entrez_genes$ENTREZID %in% "101055843",]
> length(entrez_genes$SYMBOL)
[1] 4281
因为有一些de_genes的SYMBOL没有对应的ENTREZID,因此看到少了100多个基因。
然后,把存在ENTREZID的那些基因的基因名和cluster信息提取出来
de_gene_clusters <- de_genes[de_genes$genes %in% entrez_genes$SYMBOL,
c("genes", "cluster")]
# 保持de_gene_clusters$genes的顺序不变,将他的symbol变成entrez ID
de_gene_clusters <- data.frame(
ENTREZID=entrez_genes$ENTREZID[entrez_genes$SYMBOL %in% de_gene_clusters$genes],
cluster=de_gene_clusters$cluster
)
将差异基因对应到每个cluster
也就是拆分成4个cluster组成的列表
list_de_gene_clusters <- split(de_gene_clusters$ENTREZID,
de_gene_clusters$cluster)
然后可以将4个cluster同时进行GO注释,并且放在一起(耗时!)
formula_res <- compareCluster(
ENTREZID~cluster,
data=de_gene_clusters,
fun="enrichGO",
OrgDb="org.Mm.eg.db",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05
)
# org.Mm.eg.db更新于2019年4月
# 可视化
pdf('DEG_GO_each_cluster.pdf',width = 11,height = 6)
dotplot(formula_res, showCategory=5)
dev.off()
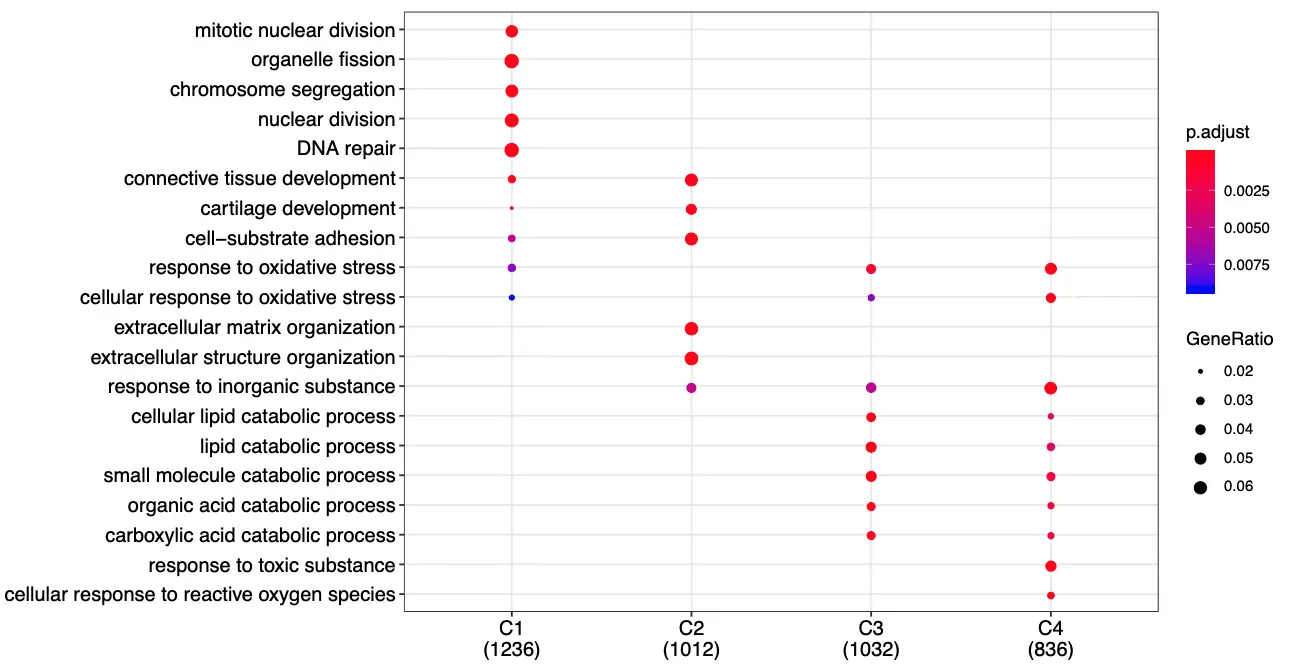
简化GO注释结果
The simplified version of enriched result is more clear and give us a more comprehensive view of the whole story https://guangchuangyu.github.io/2015/10/use-simplify-to-remove-redundancy-of-enriched-go-terms/
start_time <- Sys.time()
lineage1_ego <- simplify(
formula_res,
cutoff=0.5,
by="p.adjust",
select_fun=min
)
end_time <- Sys.time()
(end_time - start_time)
# Time difference of 1.588762 mins
pdf('DEG_GO_each_cluster_simplified.pdf',width = 11,height = 6)
dotplot(lineage1_ego, showCategory=5)
dev.off()
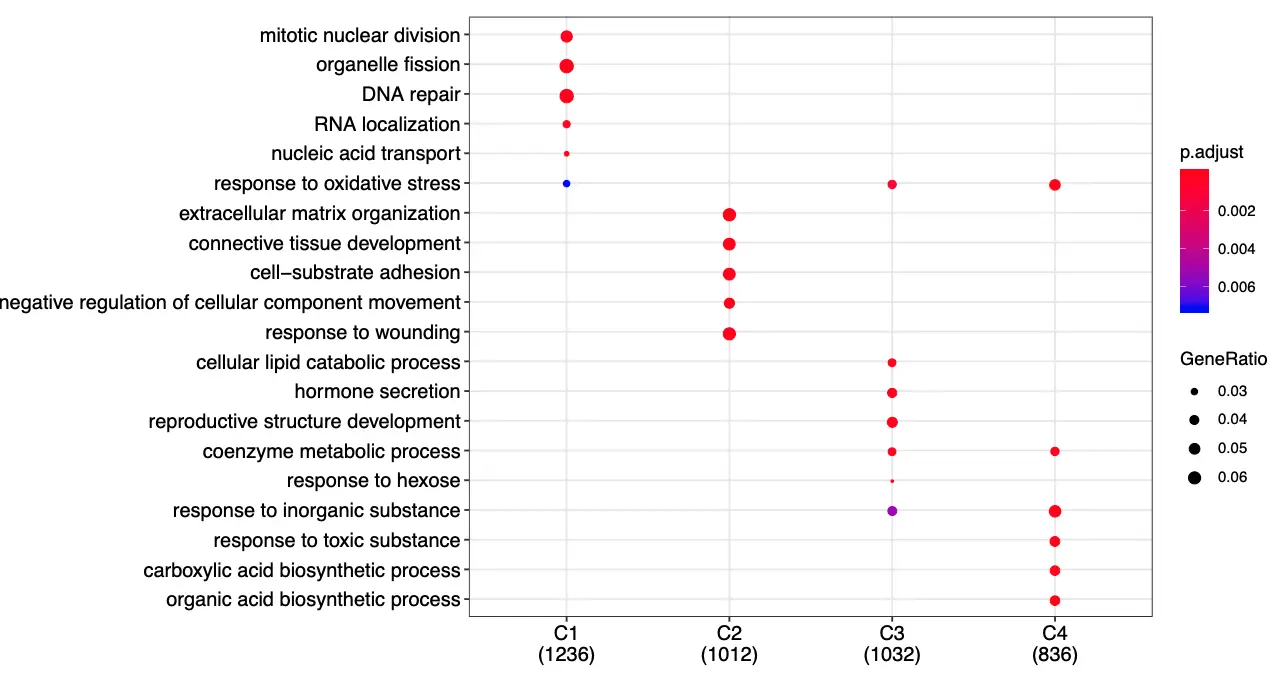
保存结果
write.csv(formula_res@compareClusterResult,
file="DEG_GO_each_cluster.csv")
# 简化版本
write.csv(lineage1_ego@compareClusterResult,
file="DEG_GO_each_cluster_simplified.csv")
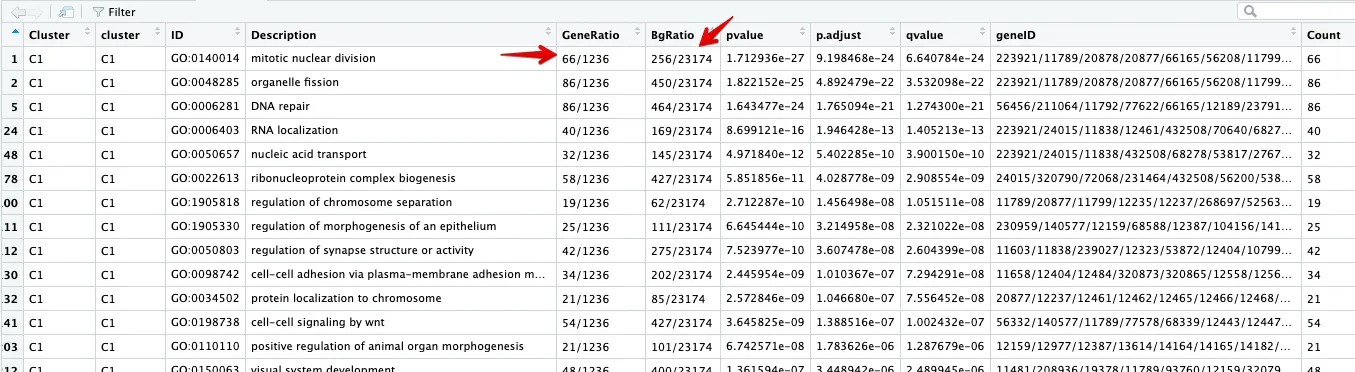
这几个数字需要好好了解一下
看第一行:背景基因是20000多个,其中属于这个GO通路的有256个基因;然后C1这个cluster这里总共得到的差异基因是1236个,其中属于这个GO通路的是66个
4 以第一个GO:0140014为例,进行探索
首先获得GO:0140014中的基因 =》 256个
library(org.Mm.eg.db)
# 结果有3百多万个
go2gene <- toTable(org.Mm.egGO2ALLEGS)
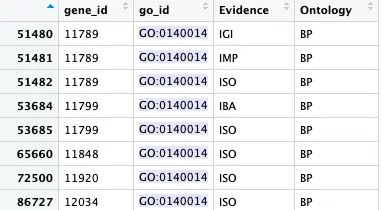
其实看到里面的基因ID存在重复,一个go_id会对应多个同样的gene_id,那么就要去重复,获取unique gene id
uni_gene <- unique(go2gene$gene_id[go2gene$go_id == 'GO:0140014'])
> length(uni_gene)
[1] 256
结果和👆的BgRatio中记录的一致
然后拿到第一群的差异基因 =》 1284个
c1_genes <- list_de_gene_clusters[['C1']]
> length(c1_genes)
[1] 1284
这里的1284比之前的1236多个几十个,说明存在几十个基因没有GO注释
找Cluster1在GO:0140014中的基因 =》 66个
其实就是找c1_genes和go2gene的交叉
overlap_genes <- intersect(c1_genes,uni_gene)
> length(overlap_genes)
[1] 66
也就是对应上面GO通路的66个基因
5 差异分析结果画热图
Monocle的结果会用到 上一篇的 1.6 用包装好的pheatmap画热图 的代码
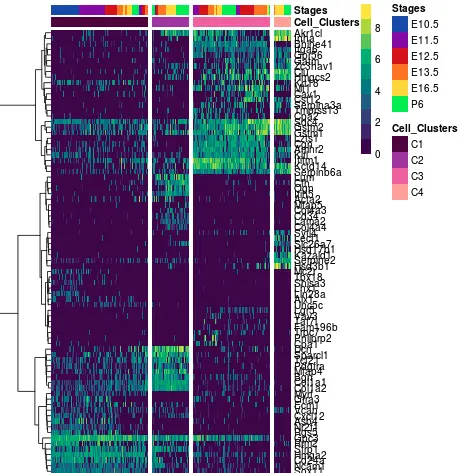
ROST的结果进行差异分析
导入数据
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
source("../analysis_functions.R")
# 加载RPKM表达矩阵
load('../female_rpkm.Rdata')
# 加载ROTS 的差异分析结果
load('DEG-ROTS_summary.Rdata')
head(summary_pop1)
head(summary_pop2)
head(summary_pop3)
head(summary_pop4)
# 6个发育时期获取
head(colnames(females))
female_stages <- sapply(strsplit(colnames(females), "_"), `[`, 1)
names(females) <- colnames(females)
table(female_stages)
# 4个cluster获取
cluster <- read.csv('female_clustering.csv')
female_clustering=cluster[,2];names(female_clustering)=cluster[,1]
table(female_clustering)
RPKM.full=females
每个群挑选各自的top18的差异基因绘制热图
population_subset<-c(rownames(summary_pop1[summary_pop1$ROTS.statistic<0,])[1:18],
rownames(summary_pop2[summary_pop2$ROTS.statistic<0,])[1:18],
rownames(summary_pop3[summary_pop3$ROTS.statistic<0,])[1:18],
rownames(summary_pop4[summary_pop4$ROTS.statistic<0,])[1:18])
总共得到了72个基因,然后取出
RPKM_heatmap<-RPKM.full[population_subset,]
将小表达矩阵的列名重新按照4个cluster排序,并且log转换
RPKM_heatmap<-RPKM_heatmap[,order(female_clustering)]
RPKM_heatmap<-log2(RPKM_heatmap+1)
每个群赋予不同颜色
popul.col<-sort(female_clustering);table(popul.col)
popul.col<-replace(popul.col, popul.col=='C1',"#1C86EE" )
popul.col<-replace(popul.col, popul.col=='C2',"#00EE00" )
popul.col<-replace(popul.col, popul.col=='C3',"#FF9912" )
popul.col<-replace(popul.col, popul.col=='C4',"#FF3E96" )
画图
library(gplots)
png("ROST-DEG-heatmap.png")
heatmap.2(as.matrix(RPKM_heatmap),
ColSideColors = as.character(popul.col), tracecol = NA,
dendrogram = "none",col=bluered, labCol = FALSE,
scale="none", key = TRUE, symkey = F, symm=F, key.xlab = "",
key.ylab = "", density.info = "density",
key.title = "log2(RPKM+1)", keysize = 1.2, denscol="black", Colv=FALSE)
dev.off()

6 两种差异分析方法结果比较
首先还是加载它们的结果
rm(list=ls())
options(stringsAsFactors = F)
load(file = 'DEG-monocle_summary.Rdata')
load(file = 'DEG-ROTS_summary.Rdata')
# monocle差异基因
mnc_DEG <- subset(de_clusters, qval<0.05)
# ROTS差异基因
head(summary_pop1)
head(summary_pop2)
head(summary_pop3)
head(summary_pop4)
对第一群比较
g1=rownames(summary_pop1[summary_pop1$FDR>0.05,])
g2=rownames(mnc_DEG[mnc_DEG$cluster=='C1',])
> length(intersect(g1,g2));length(g1);length(g2)
[1] 194
[1] 7006
[1] 1319
看到ROTS得到了7006个差异基因,monocle得到1319个,它们共有194个。就数字来讲,ROTS挑选的有些”粗糙“,一般差异基因都不会挑选太多
## 对第二群比较
g1=rownames(summary_pop2[summary_pop2$FDR>0.05,])
g2=rownames(mnc_DEG[mnc_DEG$cluster=='C2',])
> length(intersect(g1,g2));length(g1);length(g2)
[1] 620
[1] 7015
[1] 1100
# 第二群,二者的重叠度较高
## 对第三群比较
g1=rownames(summary_pop3[summary_pop3$FDR>0.05,])
g2=rownames(mnc_DEG[mnc_DEG$cluster=='C3',])
> length(intersect(g1,g2));length(g1);length(g2)
[1] 205
[1] 7909
[1] 1129
## 对第四群比较
g1=rownames(summary_pop2[summary_pop2$FDR>0.05,])
g2=rownames(mnc_DEG[mnc_DEG$cluster=='C2',])
> length(intersect(g1,g2));length(g1);length(g2)
[1] 620
[1] 7015
[1] 1100