scRNA-小鼠发育学习笔记-3-标记基因可视化
刘小泽写于19.10.24 笔记目的:根据生信技能树的单细胞转录组课程探索Smartseq2技术及发育相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 这次会介绍如何对一些标记基因进行可视化。对应视频第三单元7-8讲
前言
将对应文章这张图(不过H这张图使用的是差异基因,是下一篇的内容;这里先用marker基因尝试一下):
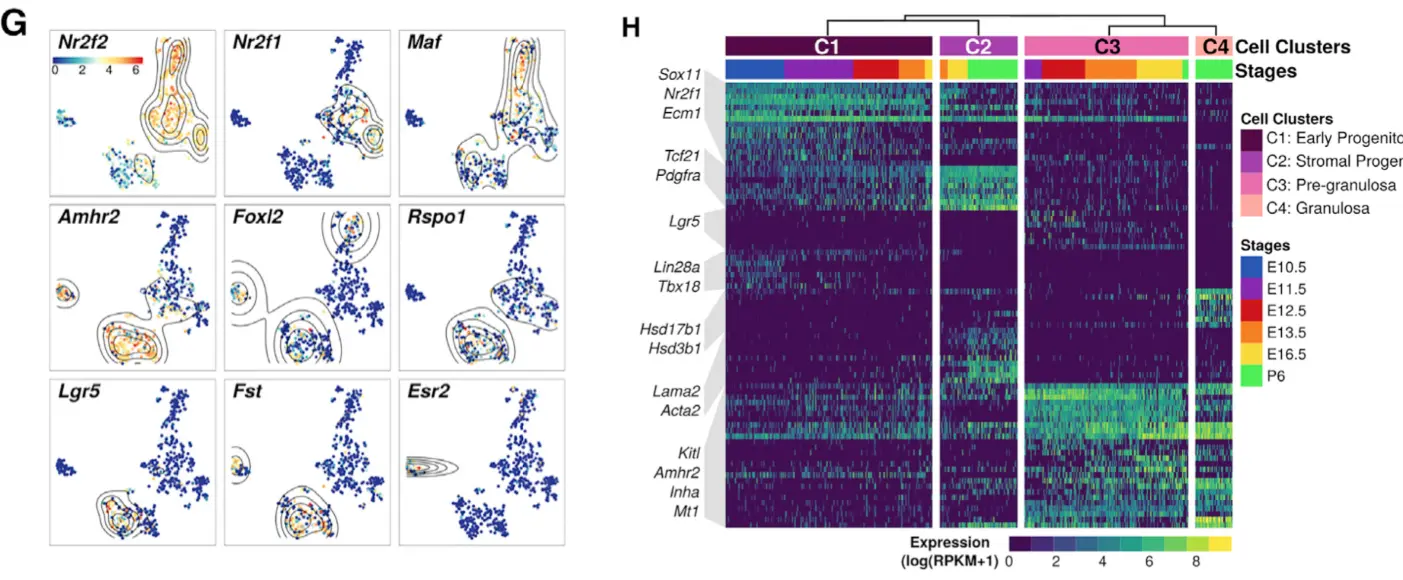
会根据之前的6个发育时期和4个cluster的tSNE结果,进行一些marker基因的等高线图和热图可视化
另外还有小提琴图:

对marker基因可视化的目的还是说明之前分的群是有道理的,文章中也提到了:

1 首先还是使用包装好的代码进行可视化
1.1 加载表达矩阵、获得cluster信息
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
source("../analysis_functions.R")
load('../female_rpkm.Rdata')
# 加载之前HCPC分群结果
cluster <- read.csv('female_clustering.csv')
female_clustering=cluster[,2];names(female_clustering)=cluster[,1]
> table(female_clustering)
female_clustering
C1 C2 C3 C4
240 90 190 43
1.2 拿到文章中的marker基因列表
作者要对哪些基因可视化,都是有自己的思量的
# 作者选择的14个marker基因
markerGenes <- c(
"Nr2f1",
"Nr2f2",
"Maf",
"Foxl2",
"Rspo1",
"Lgr5",
"Bmp2",
"Runx1",
"Amhr2",
"Kitl",
"Fst",
"Esr2",
"Amh",
"Ptges"
)
1.3 提取marker基因小表达矩阵
gene_subset <- as.matrix(log(females[rownames(females) %in% markerGenes,]+1))
> dim(gene_subset)
[1] 14 563
> gene_subset[1:4,1:4]
E10.5_XX_20140505_C01_150331_1 E10.5_XX_20140505_C02_150331_1 E10.5_XX_20140505_C03_150331_1 E10.5_XX_20140505_C04_150331_2
Kitl 2.547141 1.172887 4.1123988 4.032277
Lgr5 0.000000 0.000000 0.0568497 0.000000
Esr2 0.000000 0.000000 0.0000000 0.000000
Fst 0.000000 0.000000 0.0000000 2.694897
## 然后对这个小表达矩阵再细分,根据4个cluster的列名,也即是前面female_clustering=cluster[,2];names(female_clustering)=cluster[,1]这一步的目的
cl1_gene_subset <- gene_subset[, colnames(gene_subset) %in% names(female_clustering[female_clustering=="C1"])]
cl2_gene_subset <- gene_subset[, colnames(gene_subset) %in% names(female_clustering[female_clustering=="C2"])]
cl3_gene_subset <- gene_subset[, colnames(gene_subset) %in% names(female_clustering[female_clustering=="C3"])]
cl4_gene_subset <- gene_subset[, colnames(gene_subset) %in% names(female_clustering[female_clustering=="C4"])]
## 重新再组合起来
heatmap_gene_subset <- cbind(
cl1_gene_subset,
cl2_gene_subset,
cl3_gene_subset,
cl4_gene_subset
)
1.4 根据marker基因的顺序,重新排列这个矩阵
# 之前是这样
> rownames(heatmap_gene_subset);markerGenes
[1] "Kitl" "Lgr5" "Esr2" "Fst" "Runx1" "Amhr2" "Bmp2" "Rspo1" "Nr2f2" "Amh" "Foxl2" "Ptges" "Maf" "Nr2f1"
[1] "Nr2f1" "Nr2f2" "Maf" "Foxl2" "Rspo1" "Lgr5" "Bmp2" "Runx1" "Amhr2" "Kitl" "Fst" "Esr2" "Amh" "Ptges"
# 得到marker基因在heatmap_gene_subset中的位置
match(markerGenes,rownames(heatmap_gene_subset))
# 然后就能提取出和marker基因顺序一样的heatmap_gene_subset
heatmap_gene_subset <- heatmap_gene_subset[match(markerGenes,rownames(heatmap_gene_subset)),]
# 之后是这样
> rownames(heatmap_gene_subset);markerGenes
[1] "Nr2f1" "Nr2f2" "Maf" "Foxl2" "Rspo1" "Lgr5" "Bmp2" "Runx1" "Amhr2" "Kitl" "Fst" "Esr2" "Amh" "Ptges"
[1] "Nr2f1" "Nr2f2" "Maf" "Foxl2" "Rspo1" "Lgr5" "Bmp2" "Runx1" "Amhr2" "Kitl" "Fst" "Esr2" "Amh" "Ptges"
1.5 修改表达矩阵的列名,得到6个时间点信息
heatmap_female_stages <- sapply(strsplit(colnames(heatmap_gene_subset), "_"), `[`, 1)
> table(heatmap_female_stages)
heatmap_female_stages
E10.5 E11.5 E12.5 E13.5 E16.5 P6
68 100 103 99 85 108
1.6 用包装好的pheatmap画热图
# 看到H图中,列被分成了4栏,那么这个就是根据colbreaks来划分的。colbreaks的意思就是从哪里到哪里这是一块。当有多个分组又想画一个分割线的话,这个参数就很有用
colbreaks <- c(
ncol(cl1_gene_subset),
ncol(cl1_gene_subset)+ncol(cl2_gene_subset),
ncol(cl1_gene_subset)+ncol(cl2_gene_subset)+ncol(cl3_gene_subset)
)
# 然后就是上色,6个时间点和4个群使用自定义的颜色
cluster_color <- c(
C1="#560047",
C2="#a53bad",
C3="#eb6bac",
C4="#ffa8a0"
)
stage_color=c(
E10.5="#2754b5",
E11.5="#8a00b0",
E12.5="#d20e0f",
E13.5="#f77f05",
E16.5="#f9db21",
P6="#43f14b"
)
# 开始画热图
library(pheatmap)
png("female_marker_heatmap.png")
plot_heatmap_2(
heatmap_gene_subset,
female_clustering,
heatmap_female_stages,
rowbreaks,
colbreaks,
cluster_color,
stage_color
)
dev.off()

1.7 用包装好的ggboxplot画小提琴图
pdf("step2.1-B-markers-violin.pdf", width=10, height=22)
require(gridExtra)
# 每个基因的小提琴图都有4个cluster,对它们用不同的颜色
female_clusterPalette <- c(
"#560047",
"#a53bad",
"#eb6bac",
"#ffa8a0"
)
# 每个基因做一个小提琴图,并用for循环保存在p这个列表中
p <- list()
for (genes in markerGenes) {
p[[genes]] <- violin_gene_exp(
genes,
females,
female_clustering,
female_clusterPalette
)
}
# 最后组合起来,每列显示3张
do.call(grid.arrange,c(p, ncol=3))
dev.off()
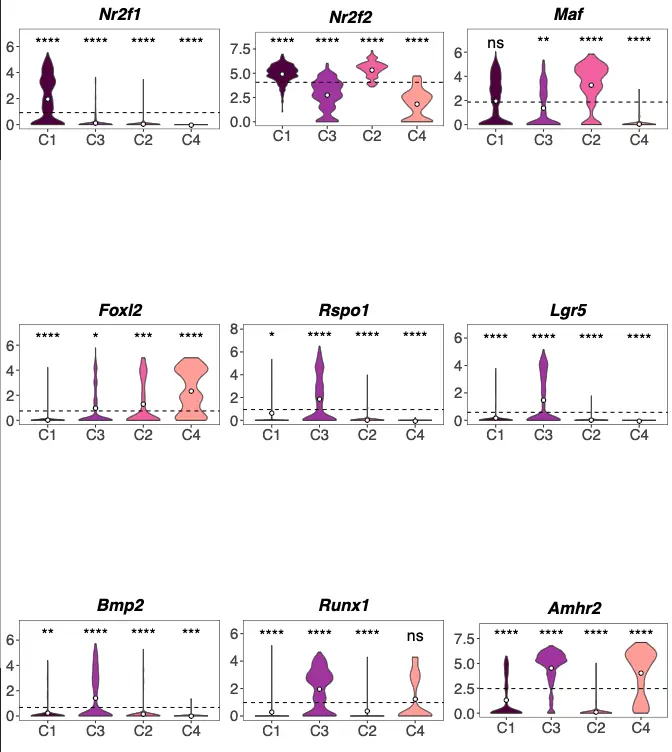
其中这个violin_gene_exp函数是精髓,如果要看它做了什么,可以按住cmd或ctrl,然后点一下这个函数,就会跳到自定义函数脚本中

1.8 用包装好的geom_point+geom_density_2d画等高线图
pdf("step2.1-C-markers-tSNE-density.pdf", width=16, height=28)
require(gridExtra)
load('../step1-female-RPKM-tSNE/female_tsne.Rdata')
p <- list()
for (genes in markerGenes) {
p[[genes]] <- tsne_gene_exp(
female_tsne,
genes,
females
)
}
do.call(grid.arrange,c(p, ncol=4))
dev.off()

2 使用Seurat包带的函数进行可视化
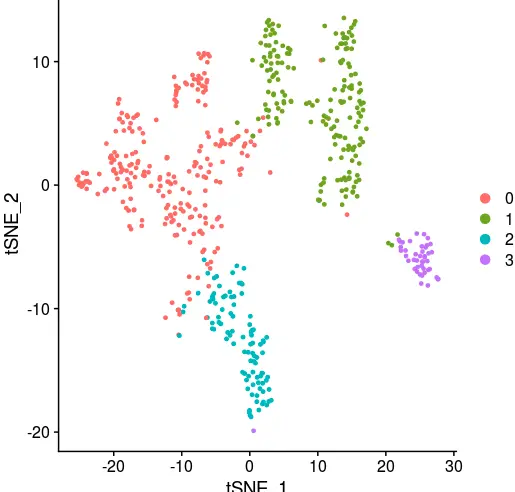
上一次已经做好了Seurat的tSNE分群结果,直接加载
load('seurat3-female-tsne.Rdata')
DimPlot(object = sce_female_tsne, reduction = "tsne")
然后画小提琴图和表达量热图
# 小提琴图
pdf('seurat3_VlnPlot.pdf', width=10, height=15)
VlnPlot(object = sce_female_tsne, features = markerGenes ,
pt.size = 0.2,ncol = 4)
dev.off()
# 基因表达量热图
pdf('seurat3_FeaturePlot.pdf', width=10, height=15)
FeaturePlot(object = sce_female_tsne, features = markerGenes ,
pt.size = 0.2,ncol = 3)
dev.off()
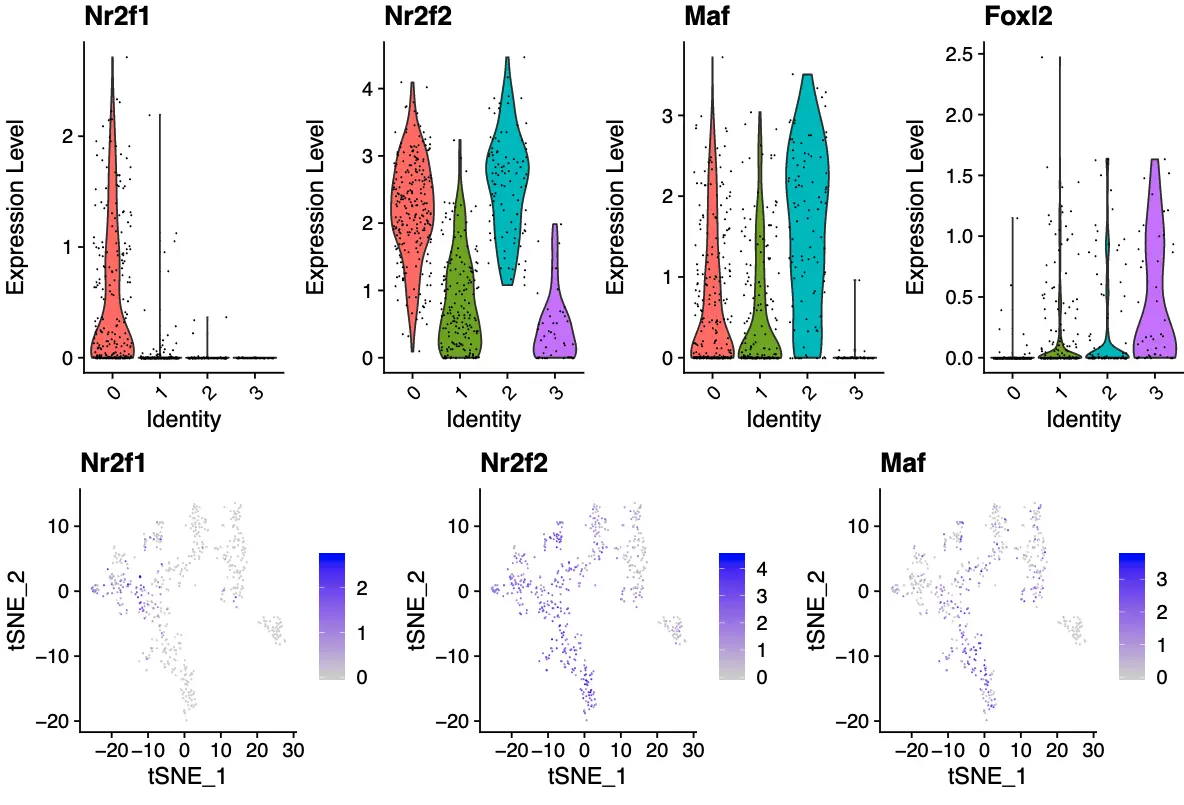
比较作者代码和Seurat的结果
取同一个基因Nr2f2,看看它们的小提琴图:

然后如果我们自己画图呢?
# 就画其中的Nr2f2基因
## 分类信息在此
group <- Seurat::Idents(sce_female)
## 表达矩阵在此
nr2f2 <- as.numeric(log(female_count['Nr2f2',]+1))
boxplot(nr2f2~group)
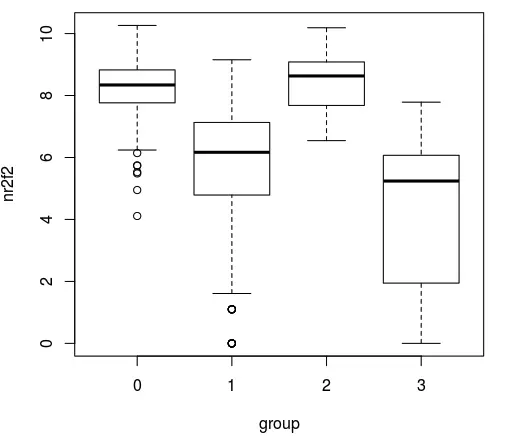
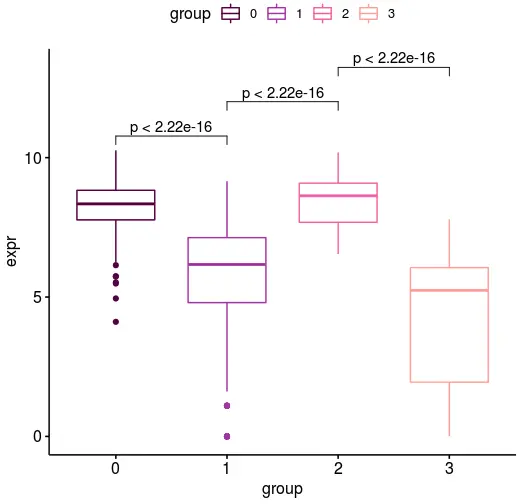
ggboxplot画一个箱线图并加上显著性
df <- data.frame(expr=nr2f2,
group=group)
female_clusterPalette <- c(
"#560047",
"#a53bad",
"#eb6bac",
"#ffa8a0"
)
my_comparisons <- list( c("0", "1"), c("1", "2"), c("2", "3") )
ggboxplot(df, x = "group", y = "expr",
color = "group", palette = female_clusterPalette)+
stat_compare_means(comparisons = my_comparisons)
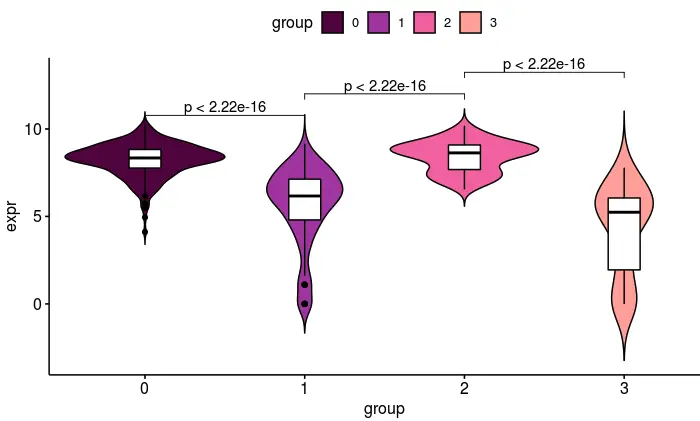
ggviolin再画一个小提琴图
df <- data.frame(expr=nr2f2,
group=group)
female_clusterPalette <- c(
"#560047",
"#a53bad",
"#eb6bac",
"#ffa8a0"
)
ggviolin(df, "group", "expr", fill = "group",
palette = female_clusterPalette,
add = "boxplot", add.params = list(fill = "white"))+
stat_compare_means(comparisons = my_comparisons)