scRNA-小鼠发育学习笔记-2-根据表达矩阵进行分群
刘小泽写于19.10.23 笔记目的:根据生信技能树的单细胞转录组课程探索Smartseq2技术及发育相关的分析 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=55 这次会介绍如何针对表达矩阵进行分群,不一定需要包装好的函数。对应视频第三单元5-6讲
前言
目的是得到这个图
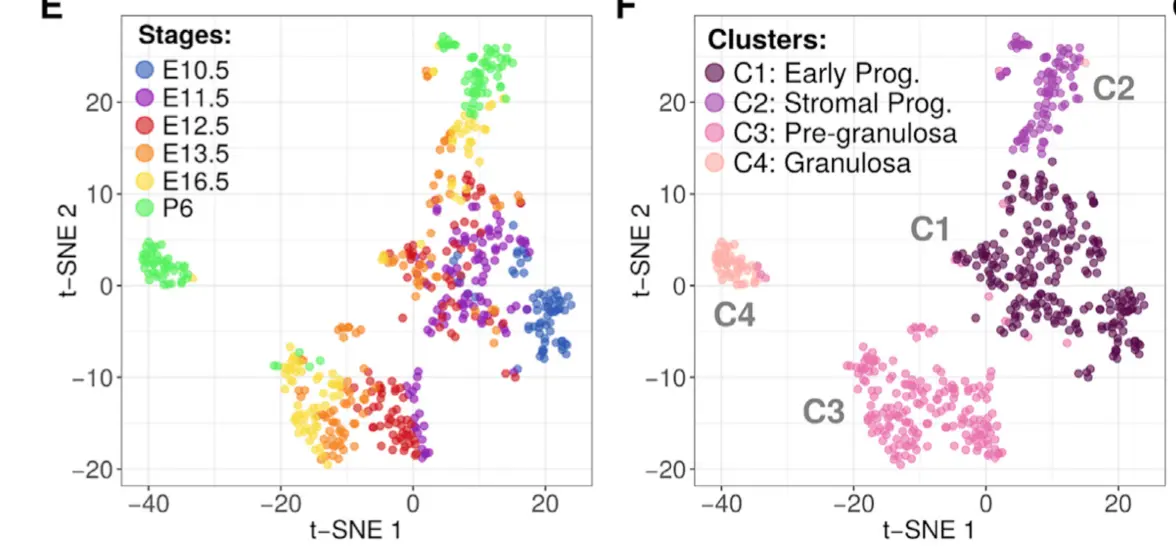
将会用到来自作者的包装好的analysis_functions.R代码:
https://github.com/IStevant/XX-XY-mouse-gonad-scRNA-seq/blob/master/scripts/analysis_functions.R
这个代码有1800多行,将会贯穿整个分析,正是这些DIY的代码,才让文章的图显得与众不同
1 首先创造表达矩阵
先下载作者上游定量处理好的数据:female_rpkm.Robj https://github.com/IStevant/XX-XY-mouse-gonad-scRNA-seq/raw/master/data/female_rpkm.Robj
一会要用的基因列表:https://github.com/IStevant/XX-XY-mouse-gonad-scRNA-seq/blob/master/data/prot_coding.csv
load(file="female_rpkm.Robj")
## 去掉重复细胞
#(例如:同一个细胞建库两次,这里作者用“rep”进行了标记)
grep("rep",colnames(female_rpkm))
colnames(female_rpkm)[256:257]
female_rpkm <- female_rpkm[,!colnames(female_rpkm) %in% grep("rep",colnames(female_rpkm), value=TRUE)]
## 只保留编码基因(去掉类似:X5430419D17Rik、BC003331等)
prot_coding_genes <- read.csv(file="prot_coding.csv", row.names=1)
females <- female_rpkm[rownames(female_rpkm) %in% as.vector(prot_coding_genes$x),]
save(females,file = 'female_rpkm.Rdata')
2 然后使用包装好的代码进行tSNE
2.1 对细胞操作=》细胞发育时期的获取
细胞是从6个时间点取出的,于是先找到这6个时间点
load('../female_rpkm.Rdata')
> dim(females)
[1] 21083 563
> head(colnames(females))
[1] "E10.5_XX_20140505_C01_150331_1" "E10.5_XX_20140505_C02_150331_1"
[3] "E10.5_XX_20140505_C03_150331_1" "E10.5_XX_20140505_C04_150331_2"
[5] "E10.5_XX_20140505_C06_150331_2" "E10.5_XX_20140505_C07_150331_3"
## 取下划线分隔的第一部分
female_stages <- sapply(strsplit(colnames(females), "_"), `[`, 1)
# 或者
female_stages <- sapply(strsplit(colnames(females), "_"),
function(x)x[1])
# 再或者
female_stages <- stringr::str_split(colnames(females),'_', simplify = T)[,1]
names(female_stages) <- colnames(females)
> table(female_stages)
female_stages
E10.5 E11.5 E12.5 E13.5 E16.5 P6
68 100 103 99 85 108
2.2 对基因操作=》基因过滤与统计
去掉在所有细胞都不表达的基因
> (dim(females))
[1] 21083 563
> females <- females[rowSums(females)>0,]
> (dim(females))
[1] 16765 563
可以看到去掉了4000多个
计算各种统计指标
# 利用apply函数对每行(每个基因)进行统计
mean_per_gene <- apply(females, 1, mean, na.rm = TRUE)
sd_per_gene <- apply(females, 1, sd, na.rm = TRUE)
mad_per_gene <- apply(females, 1, mad, na.rm = TRUE)
cv = sd_per_gene/mean_per_gene
library(matrixStats)
var_per_gene <- rowVars(as.matrix(females))
cv2=var_per_gene/mean_per_gene^2
# 存储统计结果
cv_per_gene <- data.frame(mean = mean_per_gene,
sd = sd_per_gene,
mad=mad_per_gene,
var=var_per_gene,
cv=cv,
cv2=cv2)
rownames(cv_per_gene) <- rownames(females)
head(cv_per_gene)
# 根据表达量过滤统计结果
cv_per_gene=cv_per_gene[cv_per_gene$mean>1,]
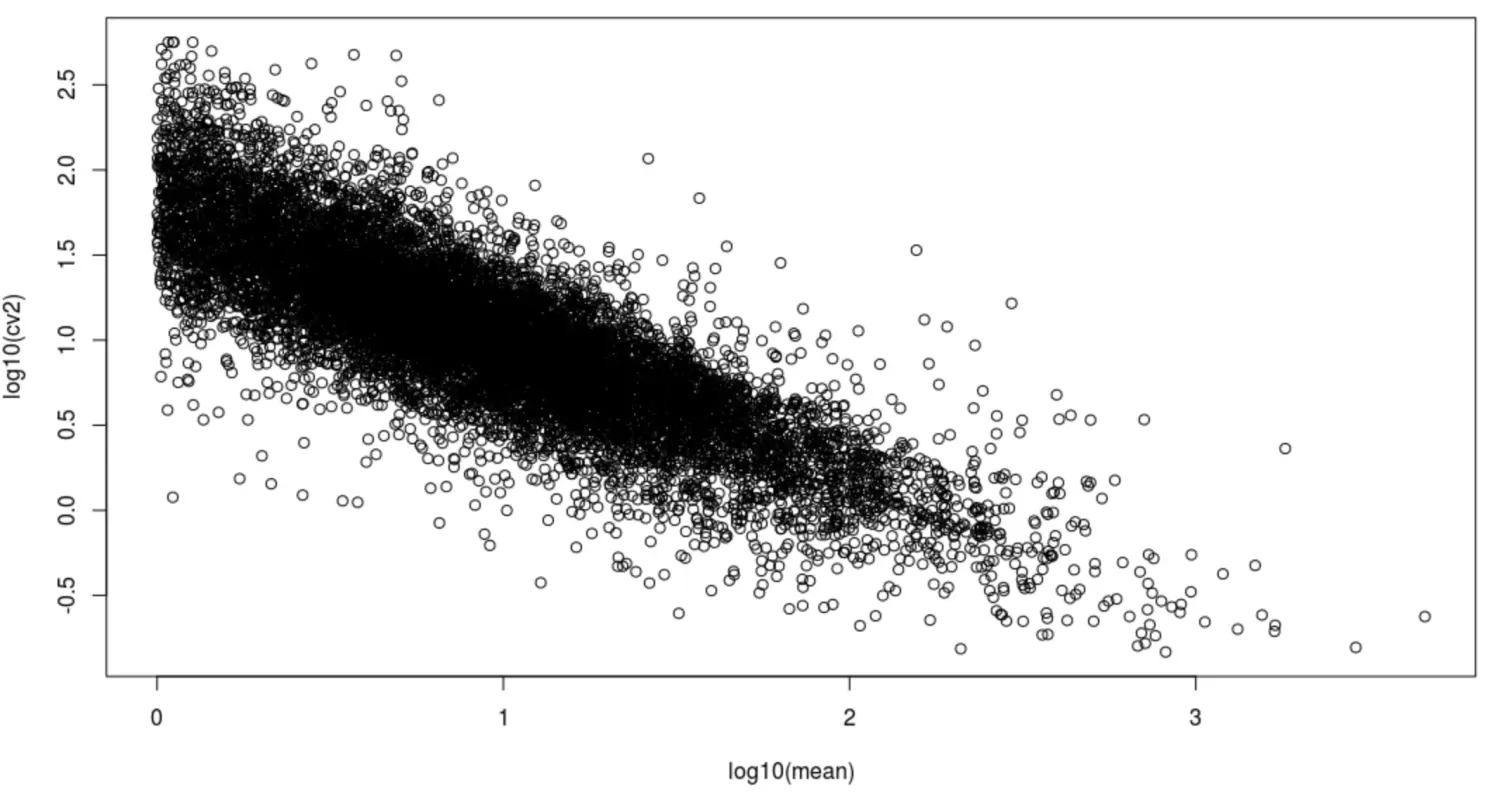
# 简易的可视化
with(cv_per_gene,plot(log10(mean),log10(cv2)))
CV值,它表示变异系数(coefficient of variation)。变异系数又称离散系数或相对偏差 ,我们肯定都知道标准偏差,也就是sd值,sd描述了数据值偏离算术平均值的程度。这个相对偏差CV描述的是标准偏差与平均值之比。
- sd值,它和均值mean、方差var一样,都是对一维数据进行的分析,如果出现两组数据测量尺度差别太大或数据量纲存在差异的话,直接用标准差就不合适了
- CV变异系数就可以解决这个问题,它利用原始数据标准差和原始数据平均值的比值来各自消除尺度与量纲的差异。
其实也可以自己构建一个简单的数据,用几行代码来证明:
mat <- matrix(c(seq(1,100),seq(1,100)*100),nrow=2,byrow = T)
dim(mat)
(sd_mat <- apply(mat, 1, sd, na.rm = TRUE))
# 29.01149 2901.14920
mean_mat <- apply(mat, 1, mean, na.rm = TRUE)
(cv_mat = sd_mat/mean_mat)
# 0.574485 0.574485
# 因此不同数量级的数据,不能直接放在一起比较变化幅度,要使用CV值
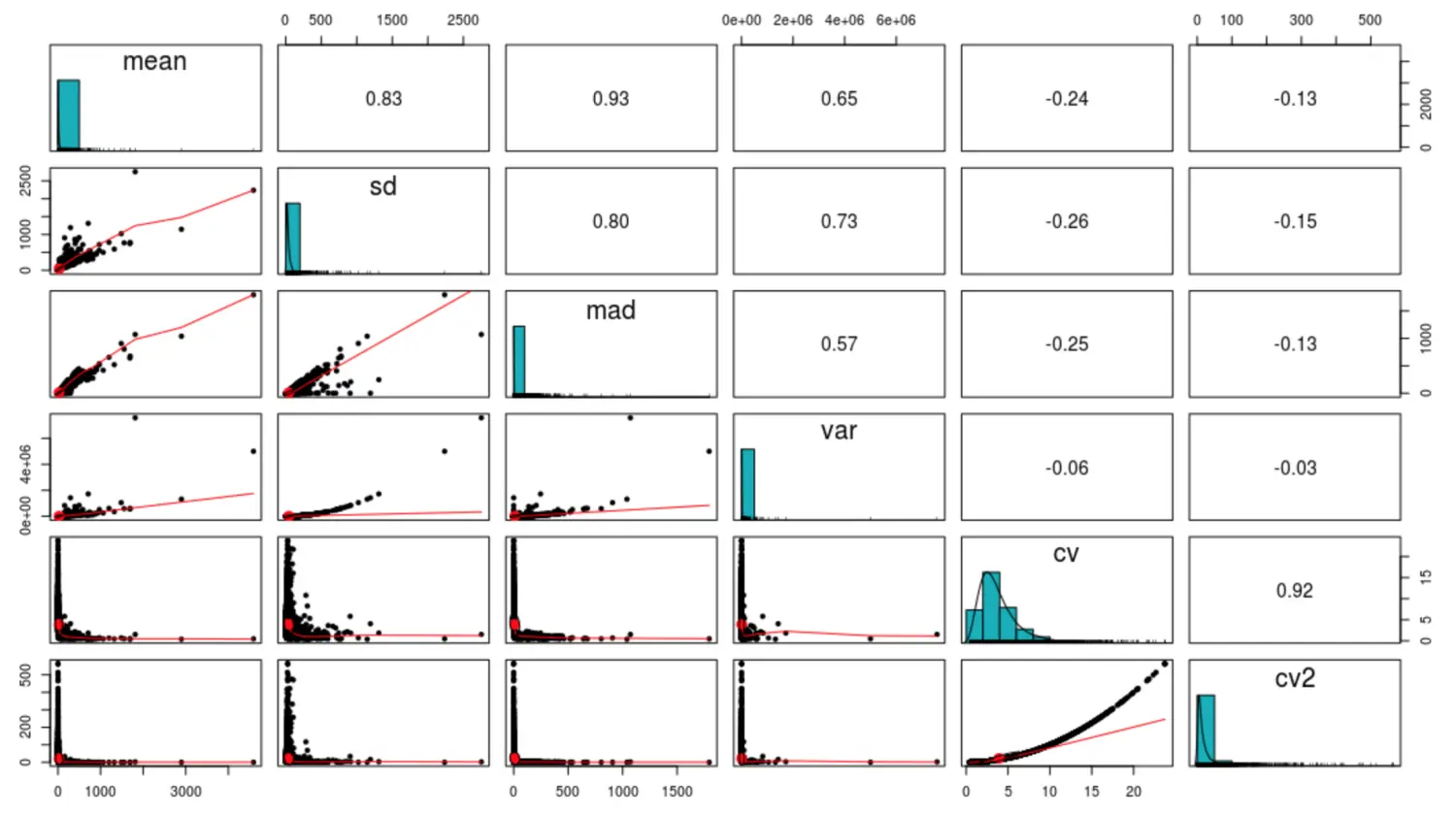
复杂一点的统计可视化:
其实就是求每列之间的相关性
library(psych)
pairs.panels(cv_per_gene,
method = "pearson", # correlation method
hist.col = "#00AFBB",
density = TRUE, # show density plots
ellipses = TRUE # show correlation ellipses
)
可以得到不同统计指标的关系
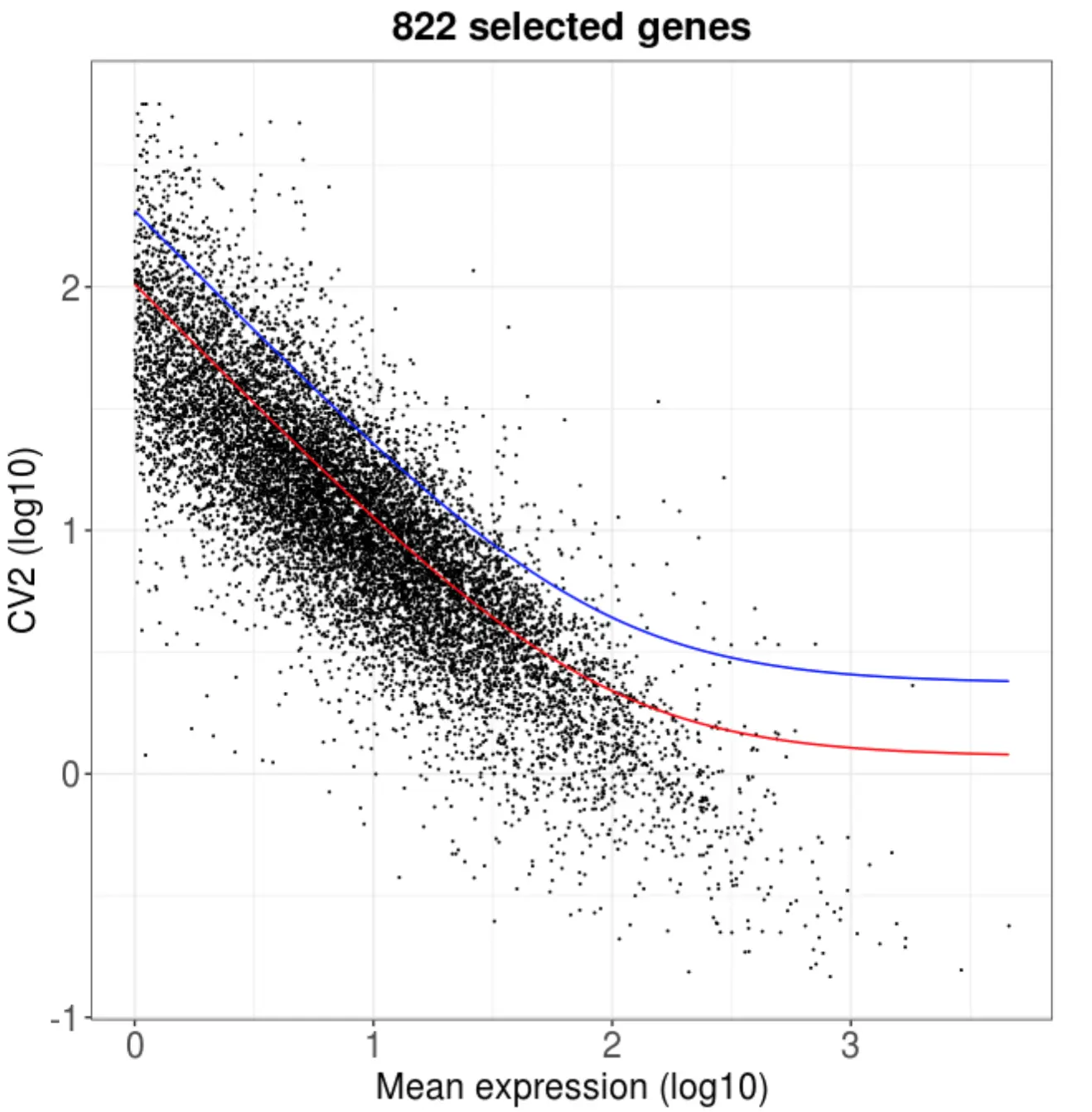
再用作者包装的函数:getMostVarGenes()
females_data <- getMostVarGenes(females, fitThr=2)
> dim(females_data)
[1] 822 563
这个函数也找了822个变化比较大的基因,用于下游分析,这其实也很像Seurat的FindVariableFeatures()做的事情
females_data <- log(females_data+1)
> females_data[1:4,1:4]
E10.5_XX_20140505_C01_150331_1 E10.5_XX_20140505_C02_150331_1
Ngfr 0 0
Slc22a18 0 0
Tspan32 0 0
Gmpr 0 0
E10.5_XX_20140505_C03_150331_1 E10.5_XX_20140505_C04_150331_2
Ngfr 0.4204863 3.619946
Slc22a18 0.0000000 0.000000
Tspan32 0.0000000 0.000000
Gmpr 0.0000000 0.000000
save(females_data,file = 'females_hvg_matrix.Rdata')
2.3 6个发育时期RtSNE分析
先是PCA
针对上面的822个HVGs进行操作
female_sub_pca <- FactoMineR::PCA(
t(females_data),
ncp = ncol(females_data),
graph=FALSE
)
然后挑选最显著的主成分,作为tSNE的输入
记得在Seurat中是使用
ElbowPlot()关注肘部的PC,这里不需要观察,直接返回最优解
significant_pcs <- jackstraw::permutationPA(
female_sub_pca$ind$coord,
B = 100,
threshold = 0.05,
verbose = TRUE,
seed = NULL
)$r
> significant_pcs
[1] 9
然后使用上面jackstraw挑出的显著主成分进行tSNE
# 6个时期给定6个颜色
female_stagePalette <- c(
"#2754b5",
"#8a00b0",
"#d20e0f",
"#f77f05",
"#f9db21",
"#43f14b"
)
female_t_sne <- run_plot_tSNE(
pca=female_sub_pca,
pc=significant_pcs,
iter=5000,
conditions=female_stages,
colours=female_stagePalette
)
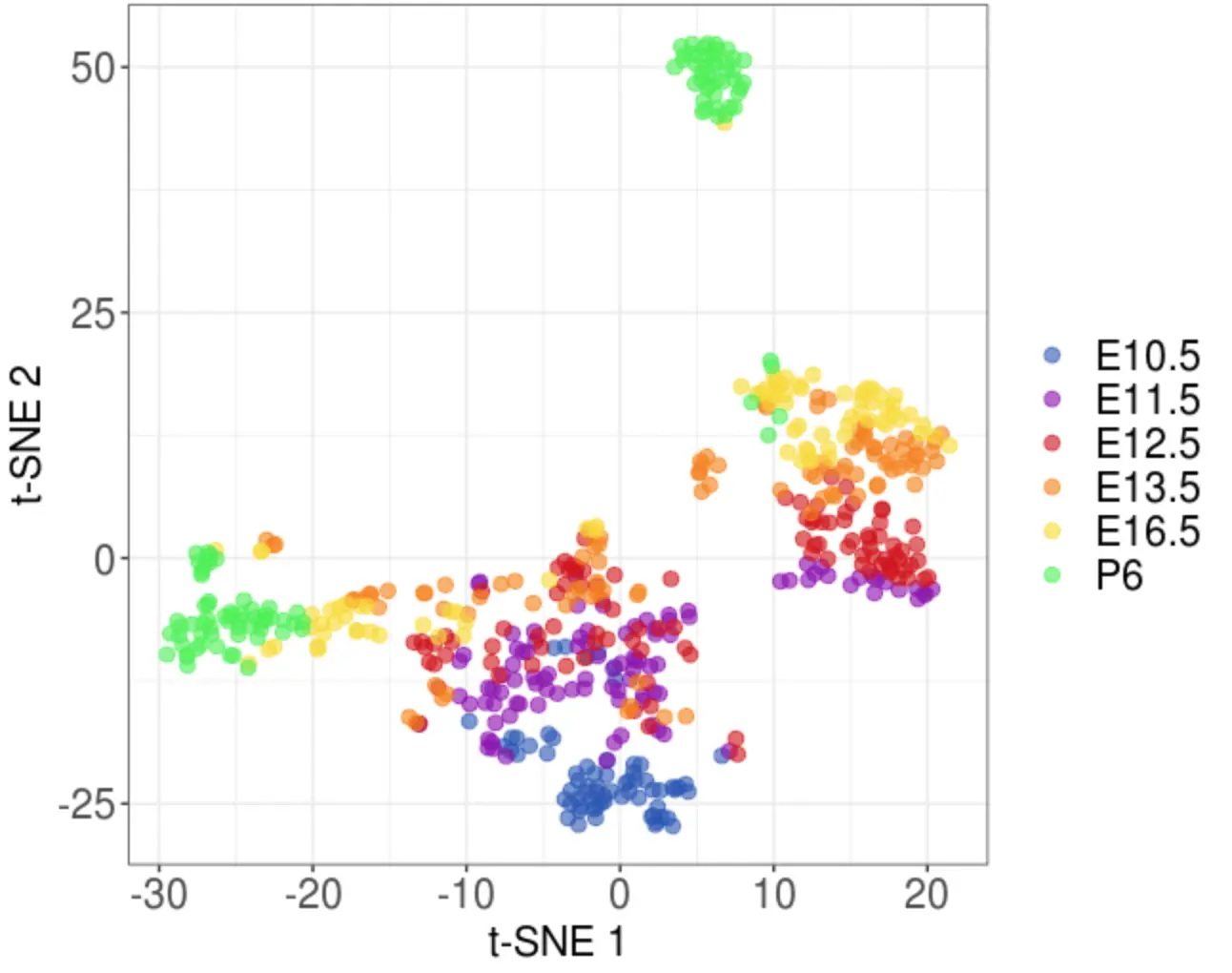
2.4 根据PCA结果进行层次聚类
采用的方法是:Hierarchical Clustering On Principle Components (HCPC)
# 使用9个显著主成分重新跑PCA
res.pca <- FactoMineR::PCA(
t(females_data),
ncp = significant_pcs,
graph=FALSE
)
# 作者根据经验认为分成4群比较好解释,于是设置4
res.hcpc <- FactoMineR::HCPC(
res.pca,
graph = FALSE,
min=4
)
# 得到分群结果
female_clustering <- res.hcpc$data.clust$clust
> table(female_clustering)
female_clustering
1 2 3 4
90 240 190 43
# 重新命名
female_clustering <- paste("C", female_clustering, sep="")
names(female_clustering) <- rownames(res.hcpc$data.clust)
# 将C1和C2调换位置
female_clustering[female_clustering=="C1"] <- "C11"
female_clustering[female_clustering=="C2"] <- "C22"
female_clustering[female_clustering=="C22"] <- "C1"
female_clustering[female_clustering=="C11"] <- "C2"
> table(female_clustering)
female_clustering
C1 C2 C3 C4
240 90 190 43
write.csv(female_clustering, file="female_clustering.csv")
还是基于之前tSNE坐标,对聚类得到的4个cluster可视化
# 为4种cluster设置颜色
female_clusterPalette <- c(
"#560047",
"#a53bad",
"#eb6bac",
"#ffa8a0"
)
> head(female_t_sne)
tSNE_1 tSNE_2 cond
E10.5_XX_20140505_C01_150331_1 -2.714291 -24.47912 E10.5
E10.5_XX_20140505_C02_150331_1 -1.580757 -26.45072 E10.5
E10.5_XX_20140505_C03_150331_1 -1.577123 -25.36753 E10.5
E10.5_XX_20140505_C04_150331_2 -6.677577 -20.00208 E10.5
E10.5_XX_20140505_C06_150331_2 3.442235 -23.32570 E10.5
E10.5_XX_20140505_C07_150331_3 3.793953 -23.33955 E10.5
# 作者包装的函数
female_t_sne_new_clusters <- plot_tSNE(
tsne=female_t_sne,
conditions=female_clustering,
colours= female_clusterPalette
)
ggsave('tSNE_cluster.pdf')
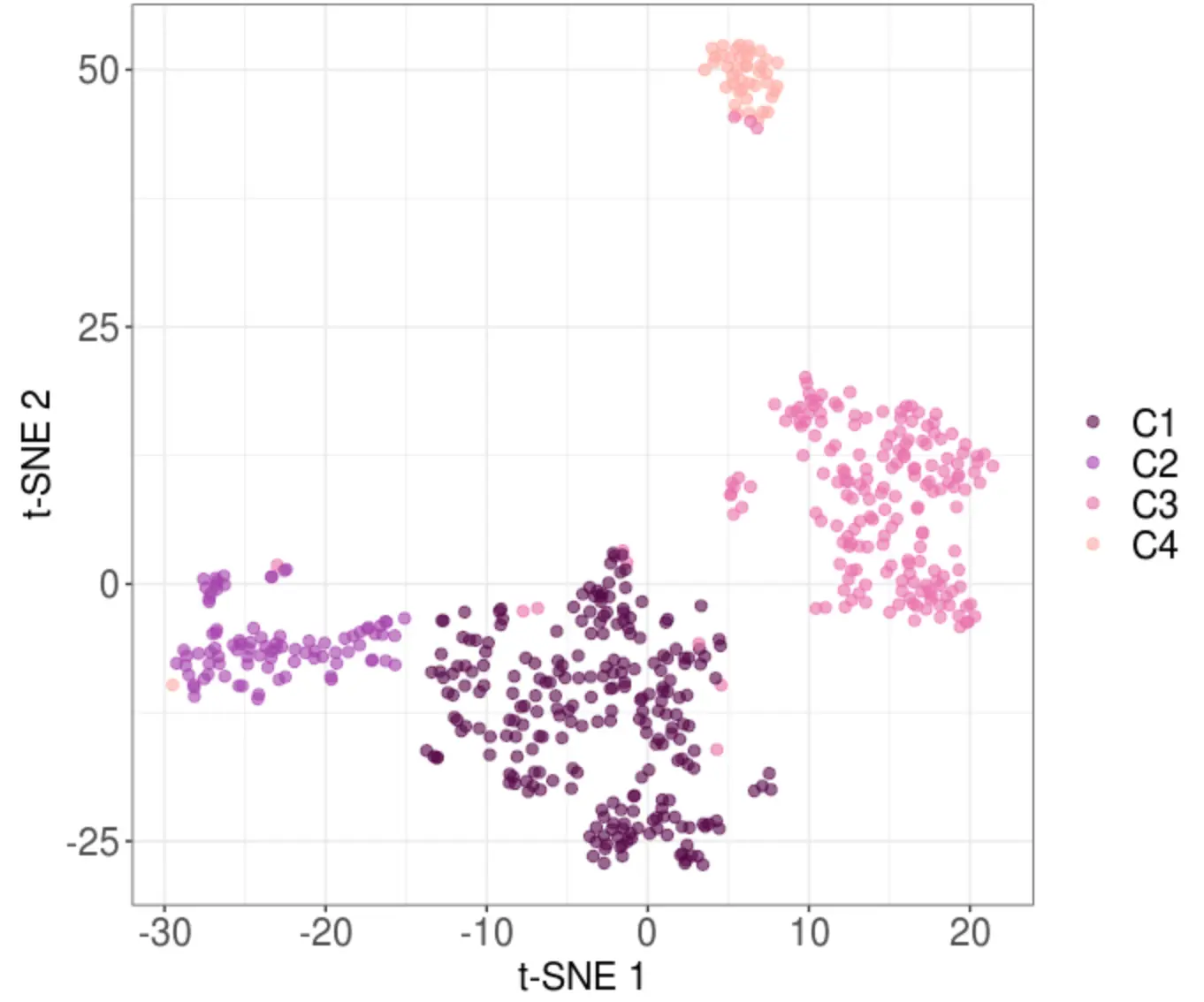
3 使用Seurat进行tSNE
上面我们使用了RPKM矩阵,下面的Seurat将会使用原始表达矩阵。当然也是推荐使用原始矩阵进行分析的
3.1 下载原始表达矩阵
链接在:https://raw.githubusercontent.com/IStevant/XX-XY-mouse-gonad-scRNA-seq/master/data/female_count.Robj
load(file="../female_count.Robj")
load('../female_rpkm.Rdata')
# 直接对细胞和基因过滤
female_count <- female_count[rownames(female_count) %in% rownames(females),!colnames(female_count) %in% grep("rep",colnames(female_count), value=TRUE)]
> female_count[1:3,1:3]
E10.5_XX_20140505_C01_150331_1 E10.5_XX_20140505_C02_150331_1
eGFP 19582 526
Gnai3 2218 122
Pbsn 0 0
E10.5_XX_20140505_C03_150331_1
eGFP 4786
Gnai3 4
Pbsn 0
save(female_count,file = '../female_count.Rdata')
3.2 对细胞操作=》细胞发育时期的获取
load('../female_count.Rdata')
female_stages <- sapply(strsplit(colnames(female_count), "_"), `[`, 1)
names(female_stages) <- colnames(female_count)
> table(female_stages)
female_stages
E10.5 E11.5 E12.5 E13.5 E16.5 P6
68 100 103 99 85 108
3.3 使用Seurat V3
构建对象
sce_female <- CreateSeuratObject(counts = female_count,
project = "sce_female",
min.cells = 1, min.features = 0)
> sce_female
An object of class Seurat
16765 features across 563 samples within 1 assay
Active assay: RNA (16765 features)
添加样本注释信息
sce_female <- AddMetaData(object = sce_female,
metadata = apply(female_count, 2, sum),
col.name = 'nUMI_raw')
sce_female <- AddMetaData(object = sce_female,
metadata = female_stages,
col.name = 'female_stages')
数据归一化
sce_female <- NormalizeData(sce_female)
sce_female[["RNA"]]@data[1:3,1:3]
找差异基因HVGs
sce_female <- FindVariableFeatures(sce_female,
selection.method = "vst",
nfeatures = 2000)
# HVGs可视化
VariableFeaturePlot(sce_female)
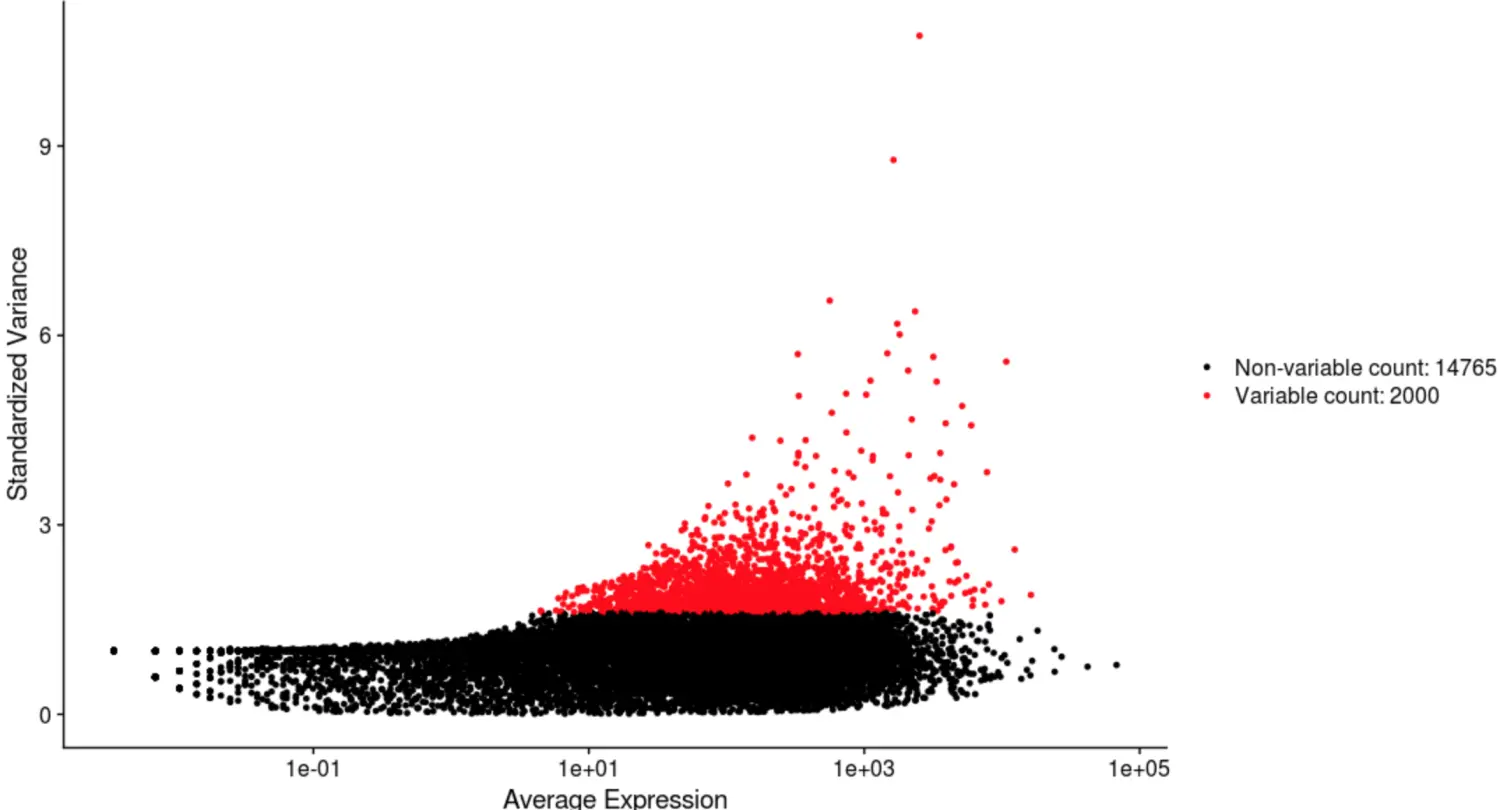
seurat3_HVGs <- VariableFeatures(sce_female)
# 检查与之前得到的HVGs重合度
load('females_hvg_matrix.Rdata')
load('seurat3_HVGs.Rdata')
length(intersect(rownames(females_data),seurat3_HVGs))
# 结果和之前822个HVGs有434个重合
数据标准化
# 默认只对FindVariableFeatures得到的HVGs进行操作
sce_female <- ScaleData(object = sce_female,
vars.to.regress = c('nUMI_raw'),
model.use = 'linear',
use.umi = FALSE)
PCA降维
sce_female <- RunPCA(sce_female,
features = VariableFeatures(object = sce_female))
降维后聚类
# 这里可以多选一些PCs
sce_female <- FindNeighbors(sce_female, dims = 1:20)
sce_female <- FindClusters(sce_female, resolution = 0.3)
进行tSNE
ElbowPlot(sce_female)
sce_female_tsne <- RunTSNE(sce_female, dims = 1:9)
tSNE结果可视化
# 6个发育时间
DimPlot(object = sce_female_tsne, reduction = "tsne",
group.by = 'female_stages')
# 4个cluster
DimPlot(sce_female_tsne, reduction = "tsne")
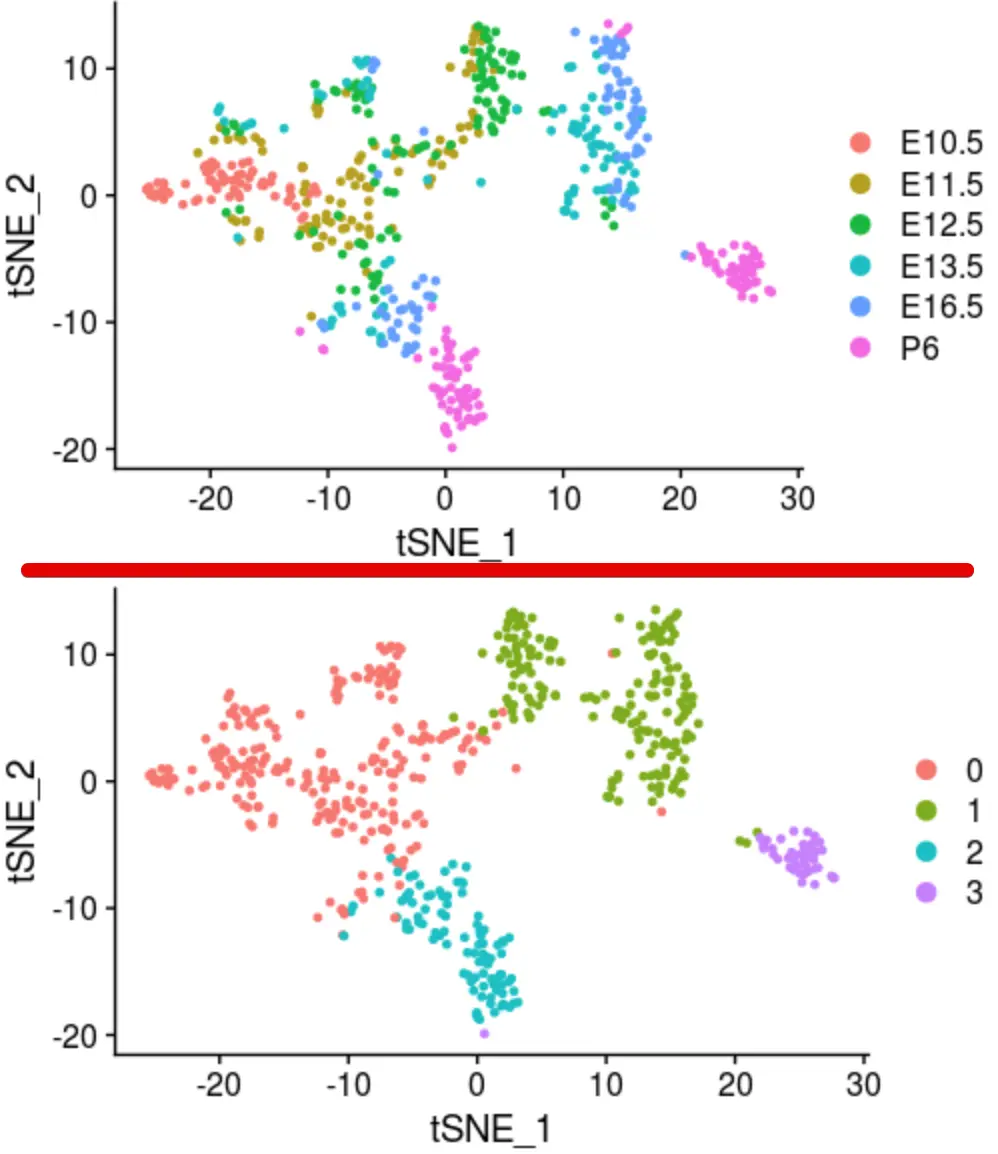
比较两次的聚类结果
cluster1 <- read.csv('female_clustering.csv')
cluster2 <- as.data.frame(Idents(sce_female_tsne))
# 把它们放在一起比较,前提条件是它们的行名相同
> identical(cluster1[,1],rownames(cluster2))
[1] TRUE
> table(cluster1[,2],cluster2[,1])
0 1 2 3
C1 224 3 13 0
C2 6 0 84 0
C3 12 177 0 1
C4 0 0 0 43
这也说明了,不同方法虽然选择的HVGs数量不同,也不完全一样,聚类的参数也不同,但最后真正的生物学意义是不会去掉的。只能说,最后选多少群是根据分析的人根据自己的理解去解释,只要参数变化,就会有各种不同的结果
4 使用更简单的函数去分群
rm(list = ls())
options(warn=-1)
options(stringsAsFactors = F)
load('../female_rpkm.Rdata')
# 根据分群获得颜色
cluster <- read.csv('female_clustering.csv')
color <- rainbow(4)[as.factor(cluster[,2])]
> table(color)
color
#00FFFFFF #8000FFFF #80FF00FF #FF0000FF
190 43 90 240
# 取前1000个sd最大的基因作为HVGs
choosed_count <- females
# 表达矩阵过滤
choosed_count <- choosed_count[apply(choosed_count, 1, sd)>0,]
choosed_count <- choosed_count[names(head(sort(apply(choosed_count, 1, sd),decreasing = T),1000)),]
进行PCA分析
pca_out <- prcomp(t(choosed_count),scale. = T)
> pca_out$x[1:3,1:3]
PC1 PC2 PC3
E10.5_XX_20140505_C01_150331_1 13.21660 -4.1600782 1.5287334
E10.5_XX_20140505_C02_150331_1 13.73109 -0.2848806 -0.8443587
E10.5_XX_20140505_C03_150331_1 10.89558 -0.2720221 -3.3839651
library(ggfortify)
autoplot(pca_out, col=color) +theme_classic()+ggtitle('PCA plot')

进行tSNE
library(Rtsne)
# 依旧选前9个
tsne_out <- Rtsne(pca_out$x[,1:9], perplexity = 10,
pca = F, max_iter = 2000,
verbose = T)
tsnes_cord <- tsne_out$Y
colnames(tsnes_cord) <- c('tSNE1','tSNE2')
ggplot(tsnes_cord, aes(x=tSNE1, y = tSNE2)) + geom_point(col=color) + theme_classic()+ggtitle('tSNE plot')

除了之前的HCPC和seurat分群,还可以利用DBSCAN、kmeans分群
# 这个运行会非常慢!
if(T){
library(Rtsne)
N_tsne <- 50
tsne_out <- list(length = N_tsne)
KL <- vector(length = N_tsne)
set.seed(1234)
for(k in 1:N_tsne)
{
tsne_out[[k]]<-Rtsne(t(log2(females+1)),initial_dims=30,verbose=FALSE,check_duplicates=FALSE,
perplexity=27, dims=2,max_iter=5000)
KL[k]<-tail(tsne_out[[k]]$itercosts,1)
print(paste0("FINISHED ",k," TSNE ITERATION"))
}
names(KL) <- c(1:N_tsne)
opt_tsne <- tsne_out[[as.numeric(names(KL)[KL==min(KL)])]]$Y
}
# DBSCAN结果
library(dbscan)
plot(opt_tsne, col=dbscan(opt_tsne,eps=3.1)$cluster,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
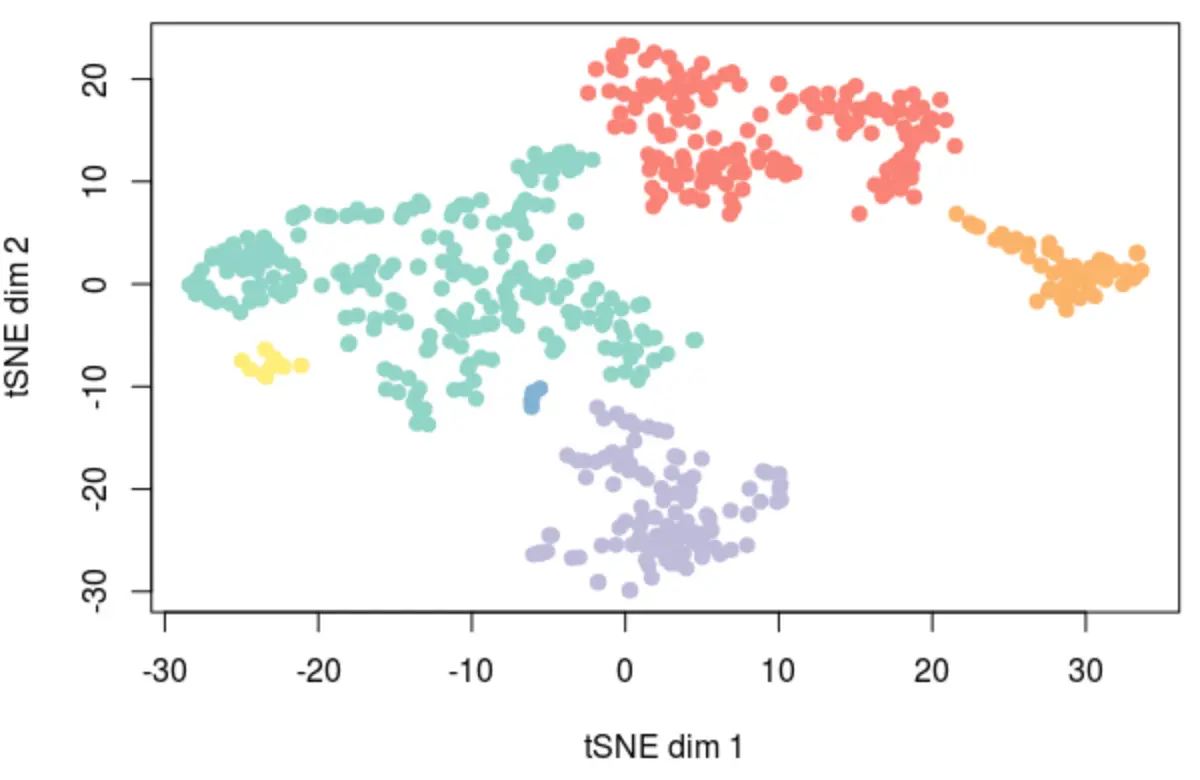
# kmeans结果
plot(opt_tsne, col=kmeans(opt_tsne,centers = 4)$clust,
pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
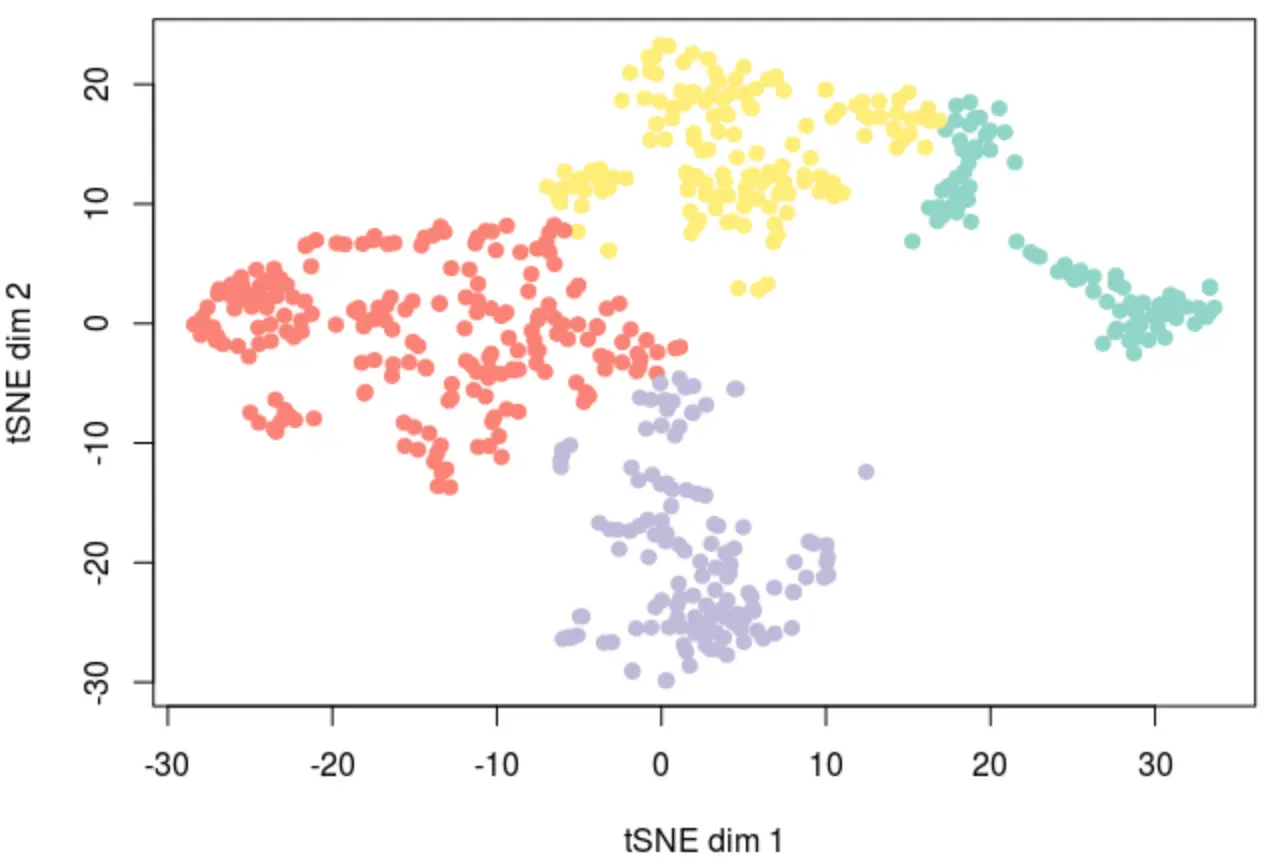
比较它们的差异
# 其中kmeans是4群
> table(kmeans(opt_tsne,centers = 4)$clust,dbscan(opt_tsne,eps=3.5)$cluster)
0 1 2 3 4
1 2 0 0 206 0
2 1 106 0 0 0
3 0 93 10 0 0
4 1 138 0 1 5