scRNA-单细胞转录组学习笔记-21-基因在任意癌症表达量相关性
刘小泽写于19.9.6-第四单元第一讲:计算基因在任意癌症表达量相关性 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
从题目可以看到,这次的主角有两个:基因和癌症中的表达量
针对第一个:我们要知道有哪些基因

从这个表中复制基因名,然后放到R中,但要注意它们中间是, 分隔,因此要使用str_split 拆分成单独的字符串:
library(stringr)
vCAF='Esam, Gng11, Higd1b, Cox4i2, Cygb, Gja4, Eng'
vCAF=unlist(str_split(vCAF,', ')) # 或者直接使用 as.character(str_split(vCAF, ', '))
mCAF='Dcn, Col12a1, Mmp2, Lum, Mrc2, Bicc1, Lrrc15, Mfap5, Col3A1, Mmp14, Spon1, Pdgfrl, Serpinf1, Lrp1, Gfpt2, Ctsk, Cdh11, Itgbl1, Col6a2, Postn, Ccdc80, Lox, Vcan, Col1a1, Fbn1, Col1a2, Pdpn, Col6a1, Fstl1, Col5a2, Aebp1'
mCAF=unlist(str_split(mCAF,', '))
> vCAF
[1] "Esam" "Gng11" "Higd1b" "Cox4i2" "Cygb" "Gja4" "Eng"
> head(mCAF)
[1] "Dcn" "Col12a1" "Mmp2" "Lum" "Mrc2" "Bicc1"
看到基因名的开头大写,其余小写,就说明是小鼠的基因名
针对第二个:如何获取癌症基因表达量信息
文章对四种癌症进行了讨论:breast cancer, pancreatic ductal adenocarcinoma, lung adenocarcinoma, and renal clear cell carcinoma
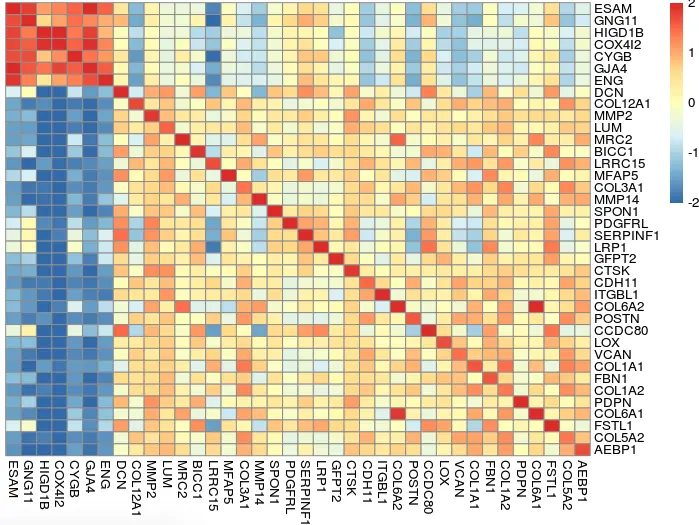
目的就是分别画这样一张图:探索两个细胞亚群(vCAF、mCAF)特有的基因在TCGA中的表现,发现两个亚群的基因都是和TCGA相关的基因在内部相关,说明了分群的效果不错

分析这张图片:这是一个相关性图,如果要做相关性的图,就要有数值型的数据,那么就是基因表达量了。我们现在有了基因名,缺的就是一个表达矩阵。因此如何获取表达矩阵就是最大的一个问题了
首先下载乳腺癌的表达矩阵
网址:https://xenabrowser.net/datapages/
但是如果从这里直接搜索BRCA的话,会有两个结果(这里选择GDC的表达矩阵):
[GDC TCGA Breast Cancer (BRCA)](https://xenabrowser.net/datapages/?cohort=GDC TCGA Breast Cancer (BRCA)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (20 datasets) 包含了6万多个基因的表达矩阵(
60,489 identifiers X 1217 samples),因为它直接使用的GenCode注释文件,不管编码与否都算作基因;基因名用的Ensembl;使用了log2(count+1)
[TCGA Breast Cancer (BRCA)](https://xenabrowser.net/datapages/?cohort=TCGA Breast Cancer (BRCA)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (30 datasets)
其中只有2万多个基因(
20,531 identifiers X 1218 samples),并且得到的是RSEM标准化表达量(使用了log2(norm_count+1));使用SYMBOL基因名
点击下图链接开始下载:文件大小133M

然后读入乳腺癌的表达矩阵
使用fread函数
library(data.table)
filepath <- file.choose()# 然后会弹出来一个对话框,找到自己下载的TCGA-BRCA.htseq_counts.tsv.gz,点OK,然后这个文件的路径就保存在了filepath
a=fread(filepath ,data.table=F)
dim(a)
# [1] 60488 1218
a[1:4,1:4]
# Ensembl_ID TCGA-E9-A1NI-01A TCGA-A1-A0SP-01A TCGA-BH-A201-01A
# 1 ENSG00000000003.13 8.787903 12.064743 11.801304
# 2 ENSG00000000005.5 0.000000 2.807355 4.954196
# 3 ENSG00000000419.11 11.054604 11.292897 11.314017
# 4 ENSG00000000457.12 10.246741 9.905387 11.117643
接着进行ID转换,Ensembl =》 Symbol ID
需要用到人类的物种注释包:org.Hs.eg.db
library(org.Hs.eg.db)
# 先看看包的简介
> org.Hs.eg.db
OrgDb object:
| DBSCHEMAVERSION: 2.1
| Db type: OrgDb
| Supporting package: AnnotationDbi
| DBSCHEMA: HUMAN_DB
| ORGANISM: Homo sapiens
| SPECIES: Human
| EGSOURCEDATE: 2019-Apr26
| EGSOURCENAME: Entrez Gene
| EGSOURCEURL: ftp://ftp.ncbi.nlm.nih.gov/gene/DATA
...
# 再看看这个注释包里有什么信息
> head(ls("package:org.Hs.eg.db"))
[1] "org.Hs.eg" "org.Hs.eg.db" "org.Hs.egACCNUM" "org.Hs.egACCNUM2EG" "org.Hs.egALIAS2EG"
[6] "org.Hs.egCHR"
# 然后看看其中Ensembl的基因是什么样子
> head(toTable(org.Hs.egENSEMBL))
gene_id ensembl_id
1 1 ENSG00000121410
2 2 ENSG00000175899
3 3 ENSG00000256069
4 9 ENSG00000171428
5 10 ENSG00000156006
6 12 ENSG00000196136
发现相对于我们得到TCGA的Emsemble ID,它没有小数点后面的部分,因此我们也需要切割Ensembl ID =>str_split()
library(stringr)
esid=str_split(a$Ensembl_ID,
'[.]',simplify = T)[,1]
> head(esid)
[1] "ENSG00000000003" "ENSG00000000005" "ENSG00000000419" "ENSG00000000457" "ENSG00000000460" "ENSG00000000938"
rownames(a)=esid
开始进行ID转换 => select()或bitr()
这里二者结果一样
# 第一种方式:官方函数
e2s=select(org.Hs.eg.db,keys = esid,columns = c( "ENSEMBL" , "SYMBOL" ),keytype = 'ENSEMBL')
dim(e2s)
# [1] 60686 2
#其中很大一部分的Ensemble ID是没有Symbol对应的。如果出去symbol为NA的值:最后也就剩下25591个基因
nrow(e2s)-sum(is.na(e2s$SYMBOL))
# [1] 25591
# 第二种方式:R包函数
library(clusterProfiler)
gene_tr <- bitr(esid, fromType = "ENSEMBL",
toType = "SYMBOL",
OrgDb = org.Hs.eg.db)
nrow(gene_tr)
# 25591
identical(e2s$SYMBOL[!is.na(e2s$SYMBOL)],gene_tr$SYMBOL)
# [1] TRUE
这样我们就同时拥有了Ensembl ID和Symbol ID:在TCGA矩阵中获取表达量用Emsembl ID,可视化用Symbol ID
# 小鼠基因变大写,然后挑出来存在于e2s的基因
vCAF=toupper(vCAF);vCAF=vCAF[vCAF %in% e2s$SYMBOL]
mCAF=toupper(mCAF);mCAF=mCAF[mCAF %in% e2s$SYMBOL]
# 得到匹配基因的Ensembl ID(总共38个基因),准备去获取表达量
ng=e2s[match(c(vCAF,mCAF),e2s$SYMBOL),1]
mat=a[ng,]
mat=mat[,-1]
dim(mat)
# [1] 38 1217
> mat[1:4,1:4]
TCGA-E9-A1NI-01A TCGA-A1-A0SP-01A TCGA-BH-A201-01A TCGA-E2-A14T-01A
ENSG00000149564 10.279611 10.059344 10.907642 10.458407
ENSG00000127920 9.776433 9.726218 10.948367 10.496854
ENSG00000131097 5.614710 4.857981 5.930737 6.658211
ENSG00000131055 6.022368 6.129283 6.629357 6.475733
最后就是计算相关性,准备绘制热图
计算相关性就是利用cor()函数,但是有个问题,它是对行处理还是对列处理?
# 都不用去搜索,自己随便测试一下就能出来结果(新建一个矩阵,然后对它进行默认的相关性分析)
> matrix(1:10,nrow = 2)
[,1] [,2] [,3] [,4] [,5]
[1,] 1 3 5 7 9
[2,] 2 4 6 8 10
> cor(matrix(1:10,nrow = 2))
[,1] [,2] [,3] [,4] [,5]
[1,] 1 1 1 1 1
[2,] 1 1 1 1 1
[3,] 1 1 1 1 1
[4,] 1 1 1 1 1
[5,] 1 1 1 1 1
# 很明显,这是对列进行处理
因此,我们如果想看基因之间的相关性,就将上面的mat矩阵转置一下就可以:
M=cor(t(mat))
colnames(M)=c(vCAF,mCAF)
rownames(M)=c(vCAF,mCAF)
# 然后为了避免高表达量对许多低表达量的遮盖,我们进行一个标准化处理
n=t(scale(t( M )))
n[n>2]=2
n[n< -2]= -2
pheatmap::pheatmap(n,cluster_rows = F,cluster_cols = F)
补充
在Xena数据库搜索pancreatic 会有5个数据集:
- [GDC TCGA Pancreatic Cancer (PAAD)](https://xenabrowser.net/datapages/?cohort=GDC TCGA Pancreatic Cancer (PAAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (14 datasets) 60,489 identifiers X 182 samples;log2(count+1) 下载地址:https://gdc.xenahubs.net/download/TCGA-PAAD.htseq_counts.tsv.gz (20M)
- [Pancreatic Cancer (Balagurunathan 2008)](https://xenabrowser.net/datapages/?cohort=Pancreatic Cancer (Balagurunathan 2008)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (2 datasets)
- [Pancreatic Cancer (Harada 2008)](https://xenabrowser.net/datapages/?cohort=Pancreatic Cancer (Harada 2008)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (2 datasets)
- [Pancreatic Cancer (Jones 2008)](https://xenabrowser.net/datapages/?cohort=Pancreatic Cancer (Jones 2008)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (2 datasets)
- [TCGA Pancreatic Cancer (PAAD)](https://xenabrowser.net/datapages/?cohort=TCGA Pancreatic Cancer (PAAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (25 datasets) 20,531 identifiers X 183 samples;log2(RSEM_norm_count+1) 下载地址:https://tcga.xenahubs.net/download/TCGA.PAAD.sampleMap/HiSeqV2.gz
在Xena数据库搜索lung adenocarcinoma 会有3个数据集:
- [GDC TCGA Lung Adenocarcinoma (LUAD)](https://xenabrowser.net/datapages/?cohort=GDC TCGA Lung Adenocarcinoma (LUAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (15 datasets) 60,489 identifiers X 585 samples 下载地址:https://gdc.xenahubs.net/download/TCGA-LUAD.htseq_counts.tsv.gz (63.8M)
- [Lung Adenocarcinoma (Ding 2008)](https://xenabrowser.net/datapages/?cohort=Lung Adenocarcinoma (Ding 2008)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (2 datasets)
- [TCGA Lung Adenocarcinoma (LUAD)](https://xenabrowser.net/datapages/?cohort=TCGA Lung Adenocarcinoma (LUAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (27 datasets) 20,531 identifiers X 576 samples 下载地址:https://tcga.xenahubs.net/download/TCGA.LUAD.sampleMap/HiSeqV2.gz