scRNA-单细胞转录组学习笔记-20-使用作者代码重复结果
刘小泽写于19.9.5-第三单元第十二+十三讲:使用作者代码重复结果 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53 这一篇会是代码密集型,因为原文作者的代码真的写的很长!
下载作者的Github
https://github.com/KPLab/SCS_CAF
文件布局如下:

下载好以后,需要将那两个tar.gz文件解压缩
看第二个R脚本 Processing.R
读入表达量数据
# 首先指定操作路径
Path_Main<-"~/scrna/SCS_CAF-master"
# 然后读入原始的第一个细胞板数据
plate1_raw<-read.delim(paste(Path_Main,"/SS2_15_0048/counts.tab",sep=""),header=TRUE,check.names=FALSE,sep="\t")
> plate1_raw[1:3,1:3]
gene A3 A6
1 Adora1 0 0
2 Sntg1 0 0
3 Prim2 0 0
作者这里考虑到重复基因名的问题
# 的确存在重复基因名
> length(as.character(plate1_raw$gene))
[1] 24490
> length(unique(as.character(plate1_raw$gene)))
> sum(duplicated(as.character(plate1_raw$gene)))
[1] 492
[1] 23998
# 用这个查看
as.character(plate1_raw$gene)[duplicated(as.character(plate1_raw$gene))]
# 看一下make.unique的用法
> make.unique(c("a", "a"))
[1] "a" "a.1"
# 将重复基因名变为唯一的名字
plate1_raw$gene<-make.unique(as.character(plate1_raw$gene))
> sum(duplicated(as.character(plate1_raw$gene)))
[1] 0
# 对样本重新命名
colnames(plate1_raw)[2:length(colnames(plate1_raw))]<-paste("SS2_15_0048_",colnames(plate1_raw)[2:length(colnames(plate1_raw))],sep="")
同样的,对于0049板,也是上述操作,最后将它们按照
gene这一列进行合并,并把gene转为行名
expr_raw<-merge(plate1_raw,plate2_raw,by="gene",all=TRUE)
rownames(expr_raw)<-as.character(expr_raw$gene)
expr_raw$gene<-NULL
最后计算一下dropout的比例(结果有点高):
# 计算dropout的比例
sum(expr_raw==0)/(dim(expr_raw)[1]*dim(expr_raw)[2])
# 0.8305757
读入ERCC数据
# 也是类似上面👆的操作
# plate1
plate1_raw_ercc<-read.delim(paste(Path_Main,"/SS2_15_0048/counts-ercc.tab",sep=""),header=TRUE,check.names=FALSE,sep="\t")
plate1_raw_ercc$gene<-make.unique(as.character(plate1_raw_ercc$gene))
colnames(plate1_raw_ercc)[2:length(colnames(plate1_raw_ercc))]<-paste("SS2_15_0048_",colnames(plate1_raw_ercc)[2:length(colnames(plate1_raw_ercc))],sep="")
# plate2
plate2_raw_ercc<-read.delim(paste(Path_Main,"/SS2_15_0049/counts-ercc.tab",sep=""),header=TRUE,check.names=FALSE,sep="\t")
plate2_raw_ercc$gene<-make.unique(as.character(plate2_raw_ercc$gene))
colnames(plate2_raw_ercc)[2:length(colnames(plate2_raw_ercc))]<-paste("SS2_15_0049_",colnames(plate2_raw_ercc)[2:length(colnames(plate2_raw_ercc))],sep="")
# 最后合并、计算ERCC dropout
expr_raw_ercc<-merge(plate1_raw_ercc,plate2_raw_ercc,by="gene",all=TRUE)
rownames(expr_raw_ercc)<-as.character(expr_raw_ercc$gene)
expr_raw_ercc$gene<-NULL
sum(expr_raw_ercc==0)/(dim(expr_raw_ercc)[1]*dim(expr_raw_ercc)[2])
# 0.6267691
看一下ERCC在各个细胞的表达量分布:
barplot(sort(as.numeric(colSums(expr_raw_ercc)),decreasing=TRUE),ylab="SPIKE LIBRARY SIZE",xlab="CELL INDEX")

然后做一个直方图,把一定数量的样本中ERCC表达量合并作一个bin:
hist(log2(as.numeric(colSums(expr_raw_ercc))+1),col="brown",
main="Distribution of Spike Library Sizes",
xlab="Spike Library Size",breaks=20)

将内源基因与ERCC spike-in合并
先看看分别有多少:
> print(paste0("There are ",nrow(expr_raw)," endogenous genes"))
[1] "There are 24490 endogenous genes"
> print(paste0("There are ",nrow(expr_raw_ercc)," spikes"))
[1] "There are 92 spikes"
合并起来:
all.counts.raw<-rbind(expr_raw,expr_raw_ercc)
> dim(all.counts.raw)
[1] 24582 768
然后重新计算dropout的比例:
sum(all.counts.raw==0)/(dim(all.counts.raw)[1]*dim(all.counts.raw)[2])
# 0.8298129
一共有7153个基因在所有细胞中表达量均为0:
dim(all.counts.raw[rowSums(all.counts.raw)==0,])
# 7153 768
关于原文去掉的52个细胞

作者也把这52个细胞的质控结果读入了R:
cell_QC<-read.delim(paste(Path_Main,"/qc/qc_2plates.filtered_cells.txt",sep=""),row.names=1,header=TRUE,sep="\t")
> dim(cell_QC)
[1] 52 6

在原始矩阵中也要去掉这些细胞:
rownames(cell_QC)<-gsub("__","_",rownames(cell_QC))
all.counts.raw<-subset(all.counts.raw,select=colnames(all.counts.raw)[!colnames(all.counts.raw)%in%rownames(cell_QC)])
> dim(all.counts.raw)
[1] 24582 716
过滤细胞后,重新拆分成count矩阵和ERCC矩阵:
# 得到原始count矩阵
expr_raw<-subset(expr_raw,select=colnames(expr_raw)[!colnames(expr_raw)%in%rownames(cell_QC)])
# 得到ERCC矩阵
expr_raw_ercc<-subset(expr_raw_ercc,select=colnames(expr_raw_ercc)[!colnames(expr_raw_ercc)%in%rownames(cell_QC)])
分别对count矩阵和ERCC矩阵过滤
all.counts.raw<-all.counts.raw[rowMeans(all.counts.raw)>0,]
expr_raw<-expr_raw[rowMeans(expr_raw)>=1,]
# count矩阵过滤后只剩下10835个基因
expr_raw_ercc<-expr_raw_ercc[rowMeans(expr_raw_ercc)>0,]
# ERCC也有原来的92个变成了89个

然后画CV vs Mean图
library("matrixStats")
# 首先还是计算CV值
mean_expr_raw<-as.numeric(rowMeans(expr_raw,na.rm=TRUE))
sd_expr_raw<-rowSds(as.matrix(expr_raw),na.rm=TRUE)
cv_squared_expr_raw<-(sd_expr_raw/mean_expr_raw)^2
# plot函数中使用(纵坐标~横坐标)
plot(log10(cv_squared_expr_raw)~log10(mean_expr_raw),
pch=20,cex=0.5,xlab="log10 ( mean raw count )",
ylab="log10 ( CV^2)",main="RAW COUNTS")
# 接下来添加ERCC的信息(画上红点)
mean_expr_raw_ercc<-as.numeric(rowMeans(expr_raw_ercc,na.rm=TRUE))
sd_expr_raw_ercc<-rowSds(as.matrix(expr_raw_ercc),na.rm=TRUE)
cv_squared_expr_raw_ercc<-(sd_expr_raw_ercc/mean_expr_raw_ercc)^2
points(log10(cv_squared_expr_raw_ercc)~log10(mean_expr_raw_ercc),col="red",pch=20,cex=1.5)
# 然后对ERCC添加loess拟合曲线
fit_expr_raw_ercc<-loess(log10(cv_squared_expr_raw_ercc)[is.finite(log10(mean_expr_raw_ercc))]~log10(mean_expr_raw_ercc)[is.finite(log10(mean_expr_raw_ercc))],span=1)
# 从小到大排个序
j<-order(log10(mean_expr_raw_ercc)[is.finite(log10(mean_expr_raw_ercc))])
#
lines(fit_expr_raw_ercc$fitted[j]~log10(mean_expr_raw_ercc)[is.finite(log10(mean_expr_raw_ercc))][j],col="red",lwd=3)
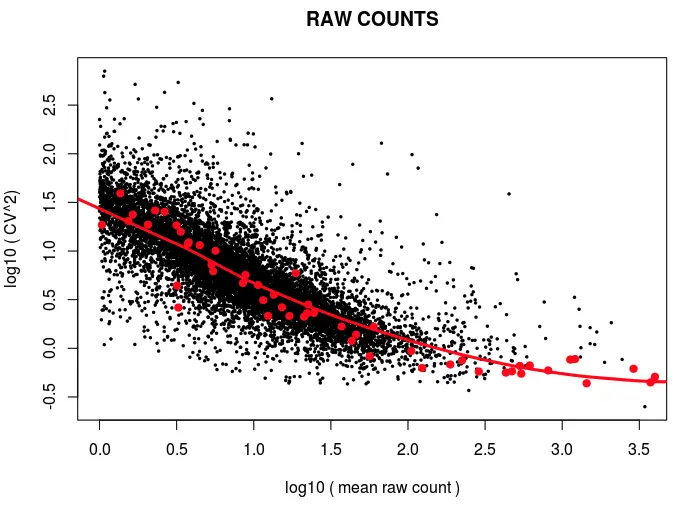
又根据拟合结果,进行了预测得到期望值,然后过滤得到符合期望CV值的基因,最后只留下5316个基因:
pred_expr_raw<-predict(fit_expr_raw_ercc,log10(mean_expr_raw))
filtered_expr_raw<-expr_raw[log10(cv_squared_expr_raw)>=pred_expr_raw,]
filtered_expr_raw<-filtered_expr_raw[grepl("NA",rownames(filtered_expr_raw))==FALSE,]
> dim(filtered_expr_raw)
[1] 5316 716
可以看到,它的过滤从原来24490的基因,然后过滤掉没表达的基因剩10835个,然后又需要符合期望,剩5000多个。最后就是拿这5000多个基因做下游分析
看第三个R脚本 Dimensionality_reduction.R
这个脚本需要RPKM结果,因此需要先跑完上面第二个完整的脚本
降维主要使用tSNE,聚类使用dbscan(它的作用和hclust或者kmeans差不多)
上来先跑50次tSNE:
library(Rtsne)
N_tsne <- 50
tsne_out <- list(length = N_tsne)
KL <- vector(length = N_tsne)
set.seed(1234)
for(k in 1:N_tsne)
{
tsne_out[[k]]<-Rtsne(t(log10(RPKM+1)),initial_dims=30,verbose=FALSE,check_duplicates=FALSE,
perplexity=27, dims=2,max_iter=5000)
KL[k]<-tail(tsne_out[[k]]$itercosts,1)
print(paste0("FINISHED ",k," TSNE ITERATION"))
}
names(KL) <- c(1:N_tsne)
# 可以看到这里选择最小的KL作为50次中效果最优的tSNE,然后主要关注tsne结果的itercosts
opt_tsne <- tsne_out[[as.numeric(names(KL)[KL==min(KL)])]]$Y
opt_tsne_full<-tsne_out[[as.numeric(names(KL)[KL==min(KL)])]]
save(tsne_out,opt_tsne,opt_tsne_full,file="step3-tsne-out.Rdata")
然后使用dbscan聚类:
library(dbscan)
plot(opt_tsne, col=dbscan(opt_tsne,eps=3.1)$cluster, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")

如果使用kmeans方法:
plot(opt_tsne, col=kmeans(opt_tsne,centers = 4)$clust, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")

看看这两种聚类方法的相关性:
> table(kmeans(opt_tsne,centers = 4)$clust,dbscan(opt_tsne,eps=3.1)$cluster)
0 1 2 3 4
1 1 226 0 0 0
2 0 0 144 0 0
3 0 27 0 0 44
4 0 231 0 43 0
# 左侧是kmeans,上方是dbscan。
发现有一个点是离群值,所以把它放到细胞数量最多的那个组:
library(dbscan)
CAFgroups<-dbscan(opt_tsne,eps=3.1)$cluster
CAFgroups_full<-dbscan(opt_tsne,eps=3.1)
CAFgroups[CAFgroups==0]<-1
CAFgroups_full$cluster[CAFgroups_full$cluster==0]<-1
plot(opt_tsne, col=CAFgroups, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")

(补充)了tSNE,还可以对PCA可视化:
CAFgroups<-dbscan(opt_tsne,eps=3.1)$cluster
CAFgroups_full<-dbscan(opt_tsne,eps=3.1)
CAFgroups[CAFgroups==0]<-1
CAFgroups_full$cluster[CAFgroups_full$cluster==0]<-1
RPKM.PCA<-prcomp(log2(t(RPKM)+1), center=TRUE)
plot(RPKM.PCA$x,main="first PCA", pch=19, col=CAFgroups)
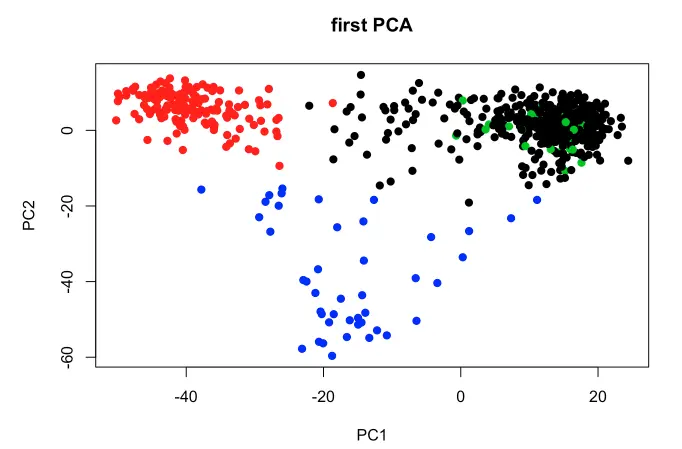
其实有了上面这个tSNE聚类图,我们就能把基因的表达量映射上去,很像Seurat的FeaturePlot()做的那样。但是这里作者自己创造函数(参考第五个脚本:Plotting.R)
需要用到基因名、表达量矩阵、tsne坐标
plot.feature2<-function(gene, tsne.output=tsne.out, DATAuse=DATA){
plot.frame<-data.frame(x=tsne.output$Y[,1], y=tsne.output$Y[,2], log2expr=as.numeric(log2(DATAuse[gene,]+1)))
p<-ggplot(plot.frame,aes(x=x, y=y, col=log2expr))+
geom_point(size=1) +
labs(title=paste(gene))+
theme_classic()+
scale_color_gradientn(colors = c("#FFFF00", "#FFD000","#FF0000","#360101"), limits=c(0,14))+
theme(axis.title = element_blank())+
theme(axis.text = element_blank())+
theme(axis.line = element_blank())+
theme(axis.ticks = element_blank())+
theme(plot.title = element_text(size=20,face="italic"))+
theme(legend.title = element_blank())+
theme(legend.position = "none")
return(p)
}
library(ggplot2)
opt_tsne <- tsne_out[[as.numeric(names(KL)[KL==min(KL)])]]$Y
opt_tsne_full<-tsne_out[[as.numeric(names(KL)[KL==min(KL)])]]
load(file='RPKM.full.Rdata')
load(file='CAFgroups.Rdata')
plot.feature2("Pdgfra", opt_tsne_full, RPKM.full)

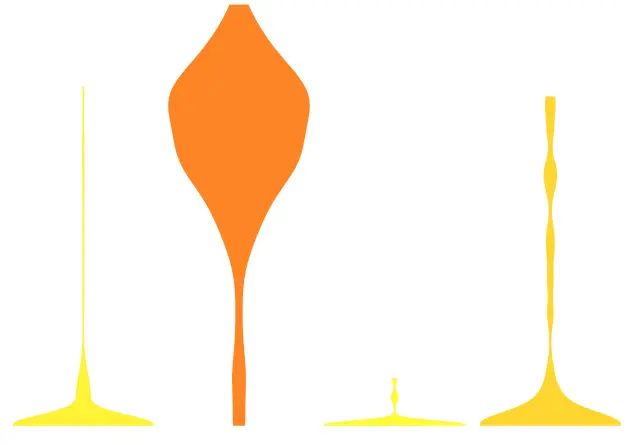
另外小提琴图的代码更是长:它是用来绘制不同基因的表达量在不同聚类分组的差异
需要用到基因名、表达量矩阵、tsne坐标
plot.violin2 <- function(gene, DATAuse, tsne.popus, axis=FALSE, legend_position="none", gene_name=FALSE){
testframe<-data.frame(expression=as.numeric(DATAuse[paste(gene),]), Population=tsne.popus$cluster)
testframe$Population <- as.factor(testframe$Population)
colnames(testframe)<-c("expression", "Population")
col.mean<-vector()
for(i in levels(testframe$Population)){
col.mean<-c(col.mean,mean(testframe$expression[which(testframe$Population ==i)]))
}
col.mean<-log2(col.mean+1)
col.means<-vector()
for(i in testframe$Population){
col.means<-c(col.means,col.mean[as.numeric(i)])
}
testframe$Mean<-col.means
testframe$expression<-log2(testframe$expression+1)
p <- ggplot(testframe, aes(x=Population, y=expression, fill= Mean, color=Mean))+
geom_violin(scale="width") +
labs(title=paste(gene), y ="log2(expression)", x="Population")+
theme_classic() +
scale_color_gradientn(colors = c("#FFFF00", "#FFD000","#FF0000","#360101"), limits=c(0,14))+
scale_fill_gradientn(colors = c("#FFFF00", "#FFD000","#FF0000","#360101"), limits=c(0,14))+
theme(axis.title.y = element_blank())+
theme(axis.ticks.y = element_blank())+
theme(axis.line.y = element_blank())+
theme(axis.text.y = element_blank())+
theme(axis.title.x = element_blank())+
theme(legend.position=legend_position )
if(axis == FALSE){
p<-p+
theme( axis.line.x=element_blank(),
axis.text.x = element_blank(),
axis.ticks.x = element_blank())
}
if(gene_name == FALSE){
p<-p+ theme(plot.title = element_blank())
}else{ p<-p + theme(plot.title = element_text(size=10,face="bold"))}
p
}
# 例如
plot.violin2(gene = "Pdgfra", DATAuse = RPKM.full, tsne.popus = CAFgroups_full)
看第四个R脚本 Differential_gene_expression.R
主要利用了ROTS包(Reproducibility-optimized test statistic),对每个亚群和其他几个亚群共同体进行比较
差异分析重点就在:表达矩阵和分组信息
library(ROTS)
library(plyr)
# 首先针对第一亚群和其他亚群比较(把其他亚群定义为234)
groups<-CAFgroups
groups[groups!=1]<-234
ROTS_input<-RPKM.full[rowMeans(RPKM.full)>=1,]
ROTS_input<-as.matrix(log2(ROTS_input+1))
# 运行代码很简单,重点就是data和group参数
results_pop1 = ROTS(data = ROTS_input, groups = groups , B = 1000 , K = 500 , seed = 1234)
# 最后根据FDR值得到第一组和其他组比较的差异基因
summary_pop1<-data.frame(summary(results_pop1, fdr=1))
head(summary_pop1)
## Row ROTS.statistic pvalue FDR
## Rgs5 8345 -19.89479 0 0
## Higd1b 4559 -17.49991 0 0
## Abcc9 393 -16.44638 0 0
## Pdpn 7193 16.02262 0 0
## Fbln2 3635 15.80534 0 0
## Rgs4 8344 -15.62123 0 0
# 同理,对第2组可以与1、3、4合并组比较;对第3组可以和第1、2、4组比较;对第4组可以和第1、2、3组比较
# 都得到以后,共同保存
save(summary_pop1,summary_pop2,summary_pop3,summary_pop4,
file = 'ROTS_summary_pop.Rdata')
每个亚群可以挑top18基因绘制热图
population_subset<-c(rownames(summary_pop1[summary_pop1$ROTS.statistic<0,])[1:18],rownames(summary_pop2[summary_pop2$ROTS.statistic<0,])[1:18],rownames(summary_pop3[summary_pop3$ROTS.statistic<0,])[1:18],rownames(summary_pop4[summary_pop4$ROTS.statistic<0,])[1:18])
RPKM_heatmap<-RPKM.full[population_subset,]
RPKM_heatmap<-RPKM_heatmap[,order(CAFgroups_full$cluster)]
RPKM_heatmap<-log2(RPKM_heatmap+1)
popul.col<-sort(CAFgroups_full$cluster)
popul.col<-replace(popul.col, popul.col==1,"#1C86EE" )
popul.col<-replace(popul.col, popul.col==2,"#00EE00" )
popul.col<-replace(popul.col, popul.col==3,"#FF9912" )
popul.col<-replace(popul.col, popul.col==4,"#FF3E96" )
library(gplots)
#pdf("heatmap_genes_population.pdf")
heatmap.2(as.matrix(RPKM_heatmap),ColSideColors = as.character(popul.col), tracecol = NA, dendrogram = "none",col=bluered, labCol = FALSE, scale="none", key = TRUE, symkey = F, symm=F, key.xlab = "", key.ylab = "", density.info = "density", key.title = "log2(RPKM+1)", keysize = 1.2, denscol="black", Colv=FALSE)

当然,原文还使用了其他几种差异分析方法,放在这里,可以做日后参考
################################
####### 第一种:DESeq2 ###########
################################
library("scran")
library("limSolve")
library(scater)
library(DESeq2)
ann<-data.frame(Plate = factor(unlist(lapply(strsplit(colnames(RPKM.full),"_"),function(x) x[3]))), Population = factor(gsub("(3|4)","2",as.character(CAFgroups)),levels=c("1","2")))
ann<-data.frame(Population = factor(gsub("(3|4)","2",as.character(CAFgroups)),levels=c("1","2")))
rownames(ann)<-colnames(RPKM.full)
ddsFullCountTable <- DESeqDataSetFromMatrix(
countData = all.counts.raw[rownames(RPKM.full),],
colData = ann,
design = ~ Population)
ddsFullCountTable<-DESeq(ddsFullCountTable)
DESeq_result<-results(ddsFullCountTable)
DESeq_result<-DESeq_result[order(DESeq_result$padj, DESeq_result$pvalue),]
head(DESeq_result,30)
write.table(DESeq_result[grep("ERCC", rownames(DESeq_result), invert=TRUE),], "DESeq_result.txt", col.names = TRUE, row.names = TRUE, quote = FALSE, sep="\t")
################################
####### 第二种:EdgeR ############
################################
library("edgeR")
edgeR_Data<-DGEList(counts=all.counts.raw[rownames(RPKM.full),], group=ann$Population)
edgeR_Data<-estimateCommonDisp(edgeR_Data)
edgeR_Data<-estimateTagwiseDisp(edgeR_Data)
edgeR_result<-exactTest(edgeR_Data)
edgeR_result_table<-edgeR_result$table
edgeR_result_table<-edgeR_result_table[order(edgeR_result_table$PValue),]
edgeR_result_table<-edgeR_result_table[grep("ERCC", rownames(edgeR_result_table), invert=TRUE),]
write.table(edgeR_result_table, "EdgeR_result.txt", col.names = TRUE, row.names = TRUE, quote=FALSE, sep="\t")
################################
###### 第三种:Wilcox ##########
################################
NumPerm<-1000
POP1_expr<-subset(RPKM.full,select=rownames(ann)[ann==1])
POP2_expr<-subset(RPKM.full,select=rownames(ann)[ann==2])
p_wilcox<-vector()
p_t<-vector()
p_perm<-vector()
statistics<-vector()
median_POP1_expr<-vector()
median_POP2_expr<-vector()
a<-seq(from=0,to=length(rownames(RPKM.full)),by=1000)
print("START DIFFERENTIAL GENE EXPRESSION BETWEEN POP1 AND POP2")
for(i in 1:length(rownames(RPKM.full)))
{
p_wilcox<-append(p_wilcox,wilcox.test(as.numeric(POP1_expr[rownames(RPKM.full)[i],]),as.numeric(POP2_expr[rownames(RPKM.full)[i],]))$p.value)
statistics<-append(statistics,wilcox.test(as.numeric(POP1_expr[rownames(RPKM.full)[i],]),as.numeric(POP2_expr[rownames(RPKM.full)[i],]))$statistic)
p_t<-append(p_t,t.test(as.numeric(POP1_expr[rownames(RPKM.full)[i],]),as.numeric(POP2_expr[rownames(RPKM.full)[i],]))$p.value)
p_perm<-append(p_perm,PermTest_Median(as.numeric(POP1_expr[rownames(RPKM.full)[i],]),as.numeric(POP2_expr[rownames(RPKM.full)[i],]),NumPerm))
median_POP1_expr<-append(median_POP1_expr,median(as.numeric(POP1_expr[rownames(RPKM.full)[i],])))
median_POP2_expr<-append(median_POP2_expr,median(as.numeric(POP2_expr[rownames(RPKM.full)[i],])))
if(i%in%a){print(paste("FINISHED ",i," GENES",sep=""))}
}
fold_change<-median_POP1_expr/median_POP2_expr
log2_fold_change<-log2(fold_change)
p_adj<-p.adjust(p_wilcox,method="fdr")
output_wilcox<-data.frame(GENE=rownames(RPKM.full),POP1_EXPR=median_POP1_expr,POP234_EXPR=median_POP2_expr,FOLD_CHANGE=fold_change,LOG2FC=log2_fold_change,WILCOX_STAT=statistics,P_T_TEST=p_t,P_PERM=p_perm,P_WILCOX=p_wilcox,FDR=p_adj)
output_wilcox<-output_wilcox[order(output_wilcox$P_PERM,output_wilcox$FDR,output_wilcox$P_WILCOX,output_wilcox$P_T_TEST,-abs(output_wilcox$LOG2FC)),]
print(head(output_wilcox,20))
write.table(output_wilcox,file="Wilcox_Perm_de_results.txt",col.names=TRUE,row.names=FALSE,quote=FALSE,sep="\t")
################################
####### 第四种:SCDE WORKFLOW
################################
library("scde")
# factor determining cell types
sg<-factor(as.numeric(CAFgroups))
# the group factor should be named accordingly
names(sg)<-colnames(expr_raw)
table(sg)
# define two groups of cells
#groups<-sg
groups <- factor(gsub("(3|4)","2",as.character(sg)),levels=c("1","2"))
table(groups)
# calculate models
cd<-apply(expr_raw,2,function(x) {storage.mode(x) <- 'integer'; x})
colnames(cd)<-colnames(expr_raw)
o.ifm<-scde.error.models(counts=cd,groups=groups,n.cores=4,threshold.segmentation=TRUE,save.crossfit.plots=FALSE,save.model.plots=FALSE,verbose=1)
print(head(o.ifm))
# filter out cells that don't show positive correlation with
# the expected expression magnitudes (very poor fits)
valid.cells<-o.ifm$corr.a > 0
table(valid.cells)
o.ifm<-o.ifm[valid.cells, ]
# estimate gene expression prior
o.prior<-scde.expression.prior(models=o.ifm,counts=cd,length.out=400,show.plot=FALSE)
# run differential expression tests on all genes.
ediff<-scde.expression.difference(o.ifm,cd,o.prior,groups=groups,n.randomizations=100,n.cores=4,verbose=1) #batch=batch
# top upregulated genes
ediff_order<-ediff[order(abs(ediff$Z),decreasing=TRUE), ]
head(ediff_order,20)
write.table(ediff_order,file="scde_de_results_1_vs_234.txt",col.names=TRUE,row.names=TRUE,quote=FALSE,sep="\t")