scRNA-单细胞转录组学习笔记-16-用Scater包分析文章数据
刘小泽写于19.8.20 第三单元第九讲:使用Scater包 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
首先再次了解文章数据
单细胞转录组学习笔记-4-获取Github代码包以及准备工作
载入数据,创建对象
rm(list = ls())
Sys.setenv(R_MAX_NUM_DLLS=999)
## 首先载入文章的数据
load(file='../input.Rdata')
# 原始表达矩阵
counts=a
counts[1:4,1:4];dim(counts) # 2万多个基因,768个细胞
library(stringr)
# 样本信息
meta=df
> head(meta)
g plate n_g all
SS2_15_0048_A3 1 0048 2624 all
SS2_15_0048_A6 1 0048 2664 all
SS2_15_0048_A5 2 0048 3319 all
SS2_15_0048_A4 3 0048 4447 all
SS2_15_0048_A1 2 0048 4725 all
SS2_15_0048_A2 3 0048 5263 all
Scater需要利用SingleCellExperiment这个对象(需要注意的是,官方友情提示,在导入对象之前,最好是将表达量数据存为矩阵)
options(warn=-1) # 全局关闭warning信息
suppressMessages(library(scater))
## 创建 scater 要求的对象
sce <- SingleCellExperiment(
assays = list(counts = as.matrix(counts)),
colData = meta
)
> sce
class: SingleCellExperiment
dim: 24582 768
metadata(0):
assays(1): counts
rownames(24582): 0610005C13Rik 0610007P14Rik ... ERCC-00170
ERCC-00171
rowData names(0):
colnames(768): SS2_15_0048_A3 SS2_15_0048_A6 ... SS2_15_0049_P22
SS2_15_0049_P24
colData names(4): g plate n_g all
reducedDimNames(0):
spikeNames(0):
预处理
如果要计算CPM值,之前一直使用log2(edgeR::cpm(dat)+1)进行计算,这个包自己做了一个函数:calculateCPM()
exprs(sce) <- log2(calculateCPM(sce ) + 1)
# 计算结果保存在
> assayNames(sce)
[1] "counts" "logcounts"
# 然后取出来计算结果也很简单
> counts(sce)[1:3,1:3]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5
0610005C13Rik 0 0 0
0610007P14Rik 0 0 18
0610009B22Rik 0 0 0
> logcounts(sce)[1:3,1:3]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5
0610005C13Rik 0 0 0.000000
0610007P14Rik 0 0 6.458664
0610009B22Rik 0 0 0.000000
注意:表达矩阵的标准命名中,
exprs和logcounts是同义词,它是为了和老版本的scater函数兼容> identical(exprs(sce), logcounts(sce)) [1] TRUE
拿到基因名,检测是否存在线粒体基因、ERCC spike-in(利用feature信息对细胞进行质控)
genes=rownames(rowData(sce))
> length(genes[grepl('^MT-',genes)])
[1] 0
> length(genes[grepl('^ERCC-',genes)])
[1] 92
# 这也是标准的ERCC,目前赛默飞公司提供的就设置了92个不同长度和GC含量的细菌RNA序列
利用calculateQCMetrics函数进行QC:
# 官方给定的写法是:
example_sce <- calculateQCMetrics(example_sce,
feature_controls = list(ERCC = 1:20, mito = 500:1000),
cell_controls = list(empty = 1:5, damaged = 31:40))
# 这里添加feature_controls信息即可
sce <- calculateQCMetrics(sce,
feature_controls = list(ERCC = grep('^ERCC',genes)))
之后过滤:
# 至少在5个细胞中有表达量的基因可以留下
keep_feature <- rowSums(exprs(sce) > 0) > 5
> table(keep_feature)
keep_feature
FALSE TRUE
10427 14155
sce <- sce[keep_feature,]
# 然后看看细胞中总表达量
boxplot(sce$total_features_by_counts )
# 根据下图设置总表达量低于2000的细胞被舍去
keep.n <- sce$total_features_by_counts > 2000
filtered_sce <- sce[,keep.n]
# 最终过滤了一万多基因,几十个细胞
> dim(filtered_sce)
[1] 14155 693
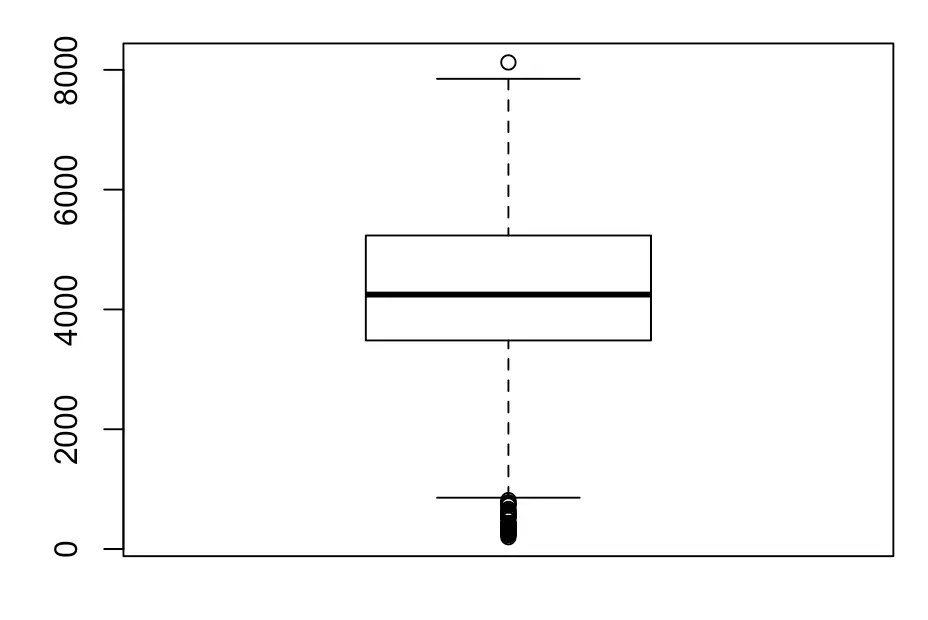
对过滤的结果可视化
首先调几个基因看看基因表达量和384孔板这个批次有没有关系:
plotExpression(sce, rownames(sce)[1:6],
x = "plate", exprs_values = "logcounts")
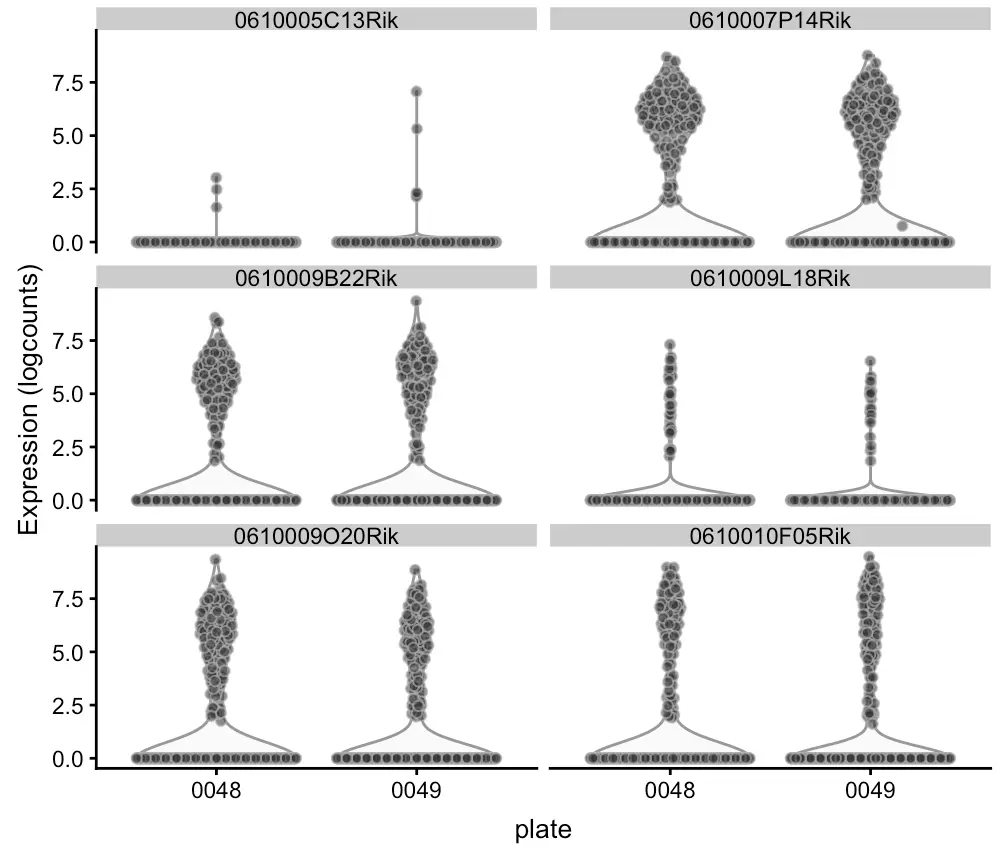
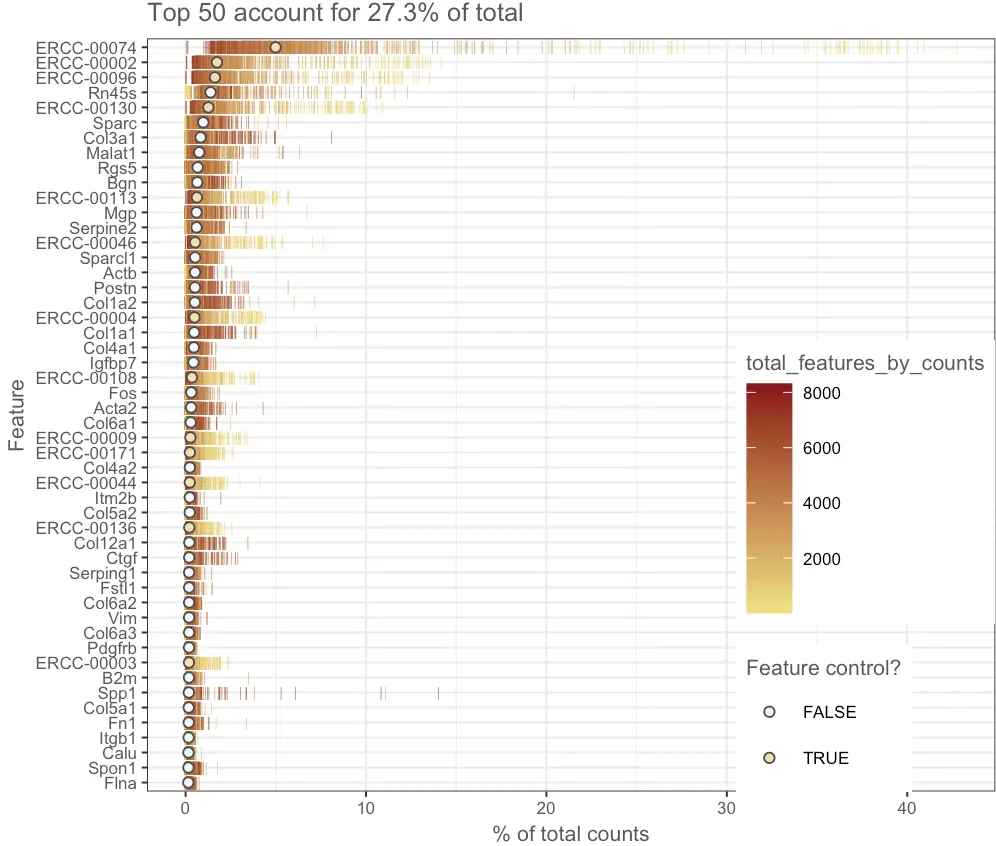
另外简单看看高表达基因的占比:
默认显示前50个基因。图中每一行表示一个基因,每个线条(bar)表示这个基因在不同细胞的表达量(可以想象成把基因表达量的箱线图转了一下)。圆圈是每个基因表达量的中位数,并且在图中经过了排序。
plotHighestExprs(sce, exprs_values = "counts")
图中可以发现top50中很多ERCC的存在,但是看到第一个ERCC在很多样本中占比超过了15%,也就是说那些样本中有超过15%的reads都”浪费“在了这种不相关的外源序列上,减少了自身的比对量
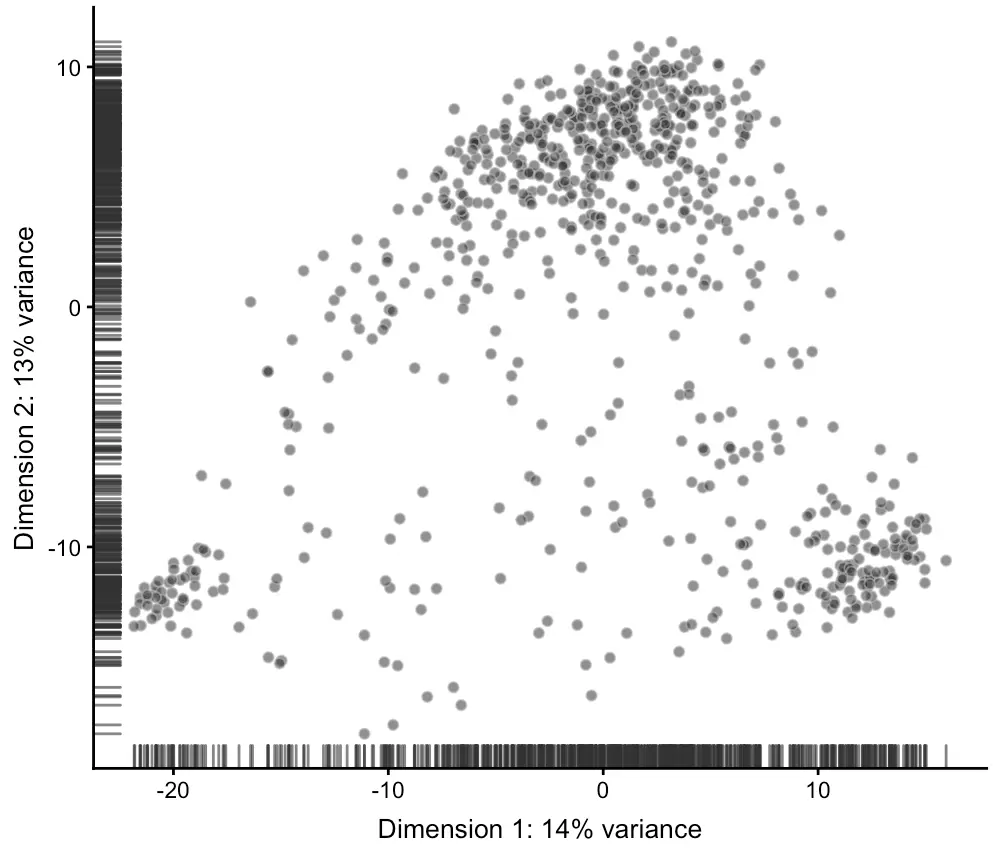
降维1-PCA分析
默认情况下,runPCA会根据500个变化差异最显著的feature的log-count值进行计算,当然这个数量可以通过ntop参数修改。
sce <- runPCA(sce)
plotPCA(sce)
# SingleCellExperiment对象中包含了reducedDims接口,其中存储了细胞降维后的坐标,可以用reducedDim、reducedDims函数获取,而具体降维的名称用reducedDimNames获取
> reducedDimNames(sce)
[1] "PCA"
这样的结果需要再加上表型信息,才能看出来是否有批次效应
plotReducedDim(sce, use_dimred = "PCA",
shape_by= "plate",
colour_by= "g")
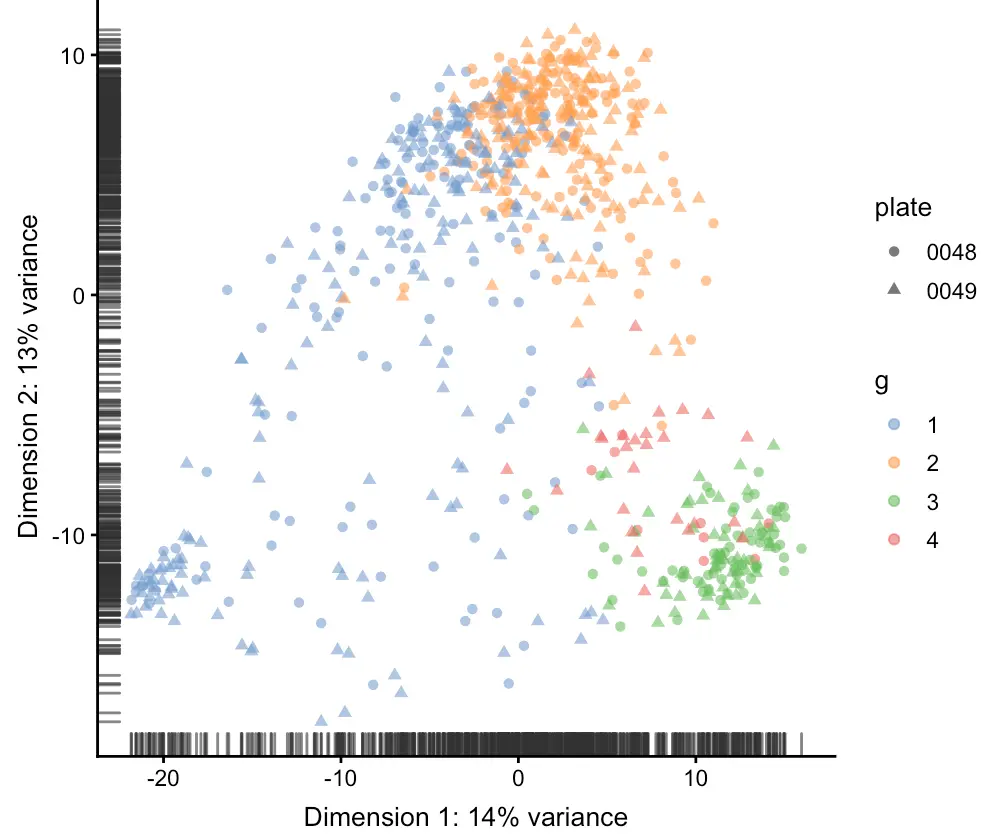
可以看到,四组层次聚类结果在PCA中也能够分开,并且每组细胞都含有两个细胞板信息,细胞板混杂在一起说明没有批次效应
降维2-tSNE
注意:tsne只是一种降维的方法,最后只给出一个坐标。根据坐标是能明白这一群和那一群能分得开,但还不能确定这一群就是单独的一组。为了找到这个依据,就是要进行聚类
set.seed(1000)
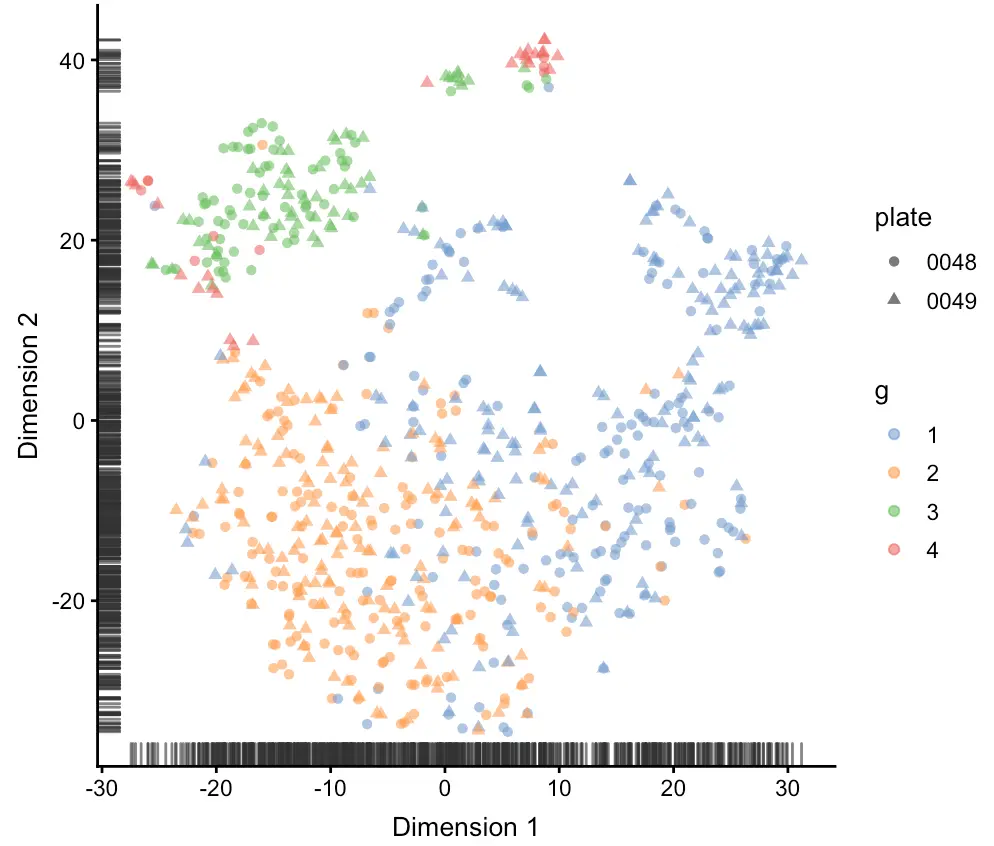
sce <- runTSNE(sce, perplexity=10)
plotTSNE(sce,
shape_by= "plate",
colour_by= "g")
从图中可以看到,之前用层次聚类定义的4群细胞,现在主要有3群(第1群和第2群混在了一起)
降维3- DiffusionMap
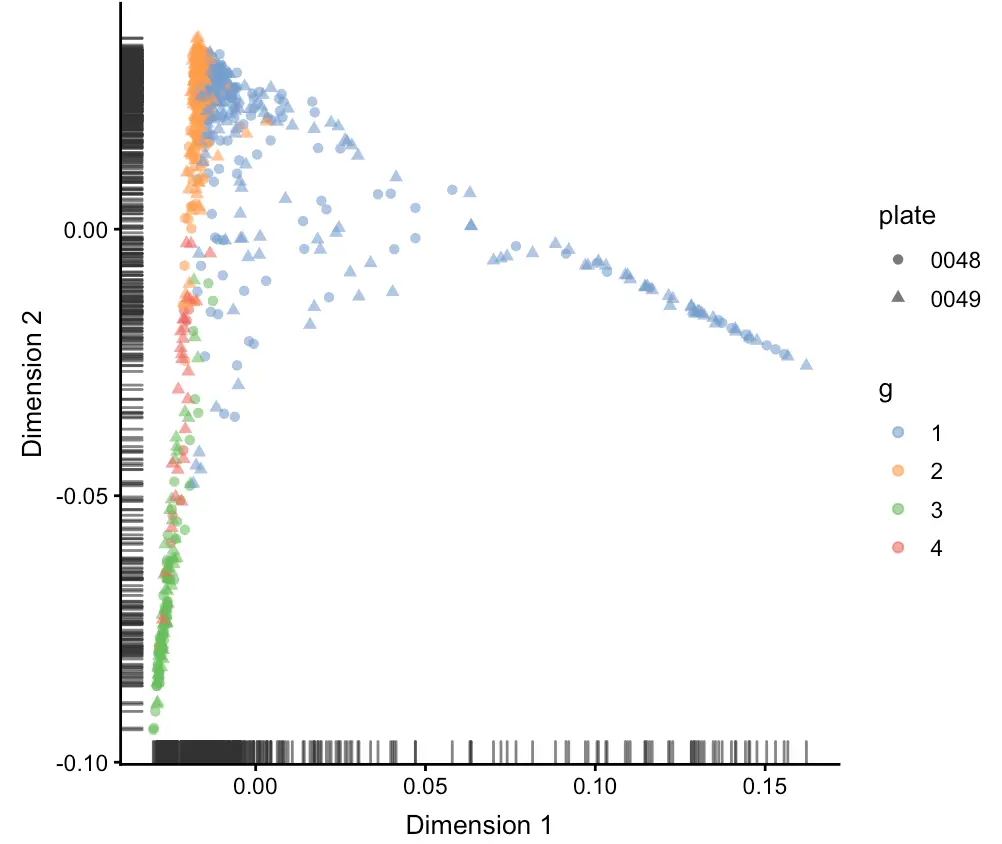
sce <- runDiffusionMap(sce)
plotDiffusionMap(sce,
shape_by= "plate",
colour_by= "g")
# 此时sce也记录了,做过三次降维处理,但每次都不冲突,存储在平行位置
> sce@reducedDims
List of length 3
names(3): PCA TSNE DiffusionMap
聚类1-K-means
以t-SNE结果为例
# 提取tsne降维后的主成分坐标,这个坐标就相当于一个新的矩阵,只不过不记录表达量而记录坐标(供后续聚类使用)
> head(sce@reducedDims$TSNE,3)
[,1] [,2]
[1,] 13.99815 -17.82133
[2,] 14.94079 -14.26358
[3,] 10.63692 -23.44417
# Kmeans需要的参数主要是:一个数值矩阵,一个自定义的分群数量
kmeans(sce@reducedDims$TSNE,centers = 4)
# 它的结果主要包含:
# Available components:
[1] "cluster" "centers" "totss" "withinss"
[5] "tot.withinss" "betweenss" "size" "iter"
[9] "ifault"
# 我们选择第一个”cluster“,就是记录的分组信息
> head(kmeans(sce@reducedDims$TSNE,centers = 4)$cluster)
[1] 4 4 4 3 1 3
# 最后将分组信息添加到sce的表型信息中
colData(sce)$tSNE_kmeans <- as.character(kmeans(sce@reducedDims$TSNE,
centers = 4)$cluster)
plotTSNE(sce, colour_by = "tSNE_kmeans")
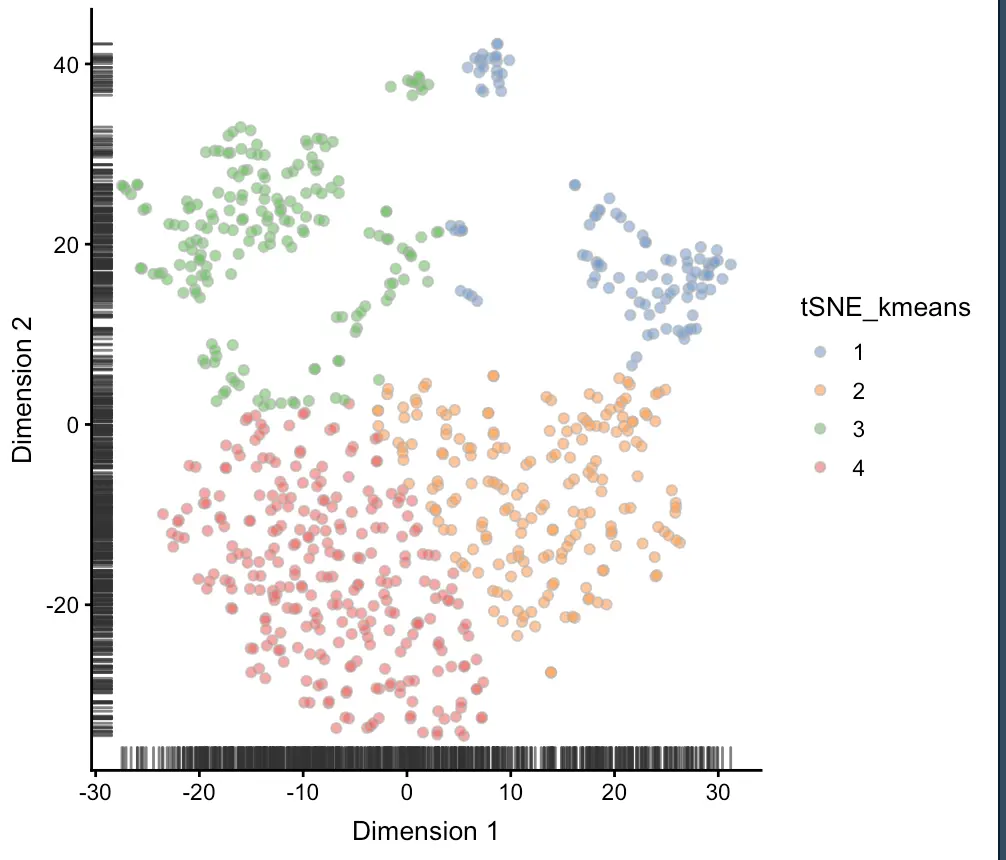
聚类2-层次聚类hclust:
# 重点:将tsne的结果当成一个矩阵即可,让其中的坐标分成4群就行
hc=hclust(dist( sce@reducedDims$TSNE ))
clus = cutree(hc, 4)
colData(sce)$tSNE_hc <- as.character(clus)
plotTSNE(sce, colour_by = "tSNE_hc")
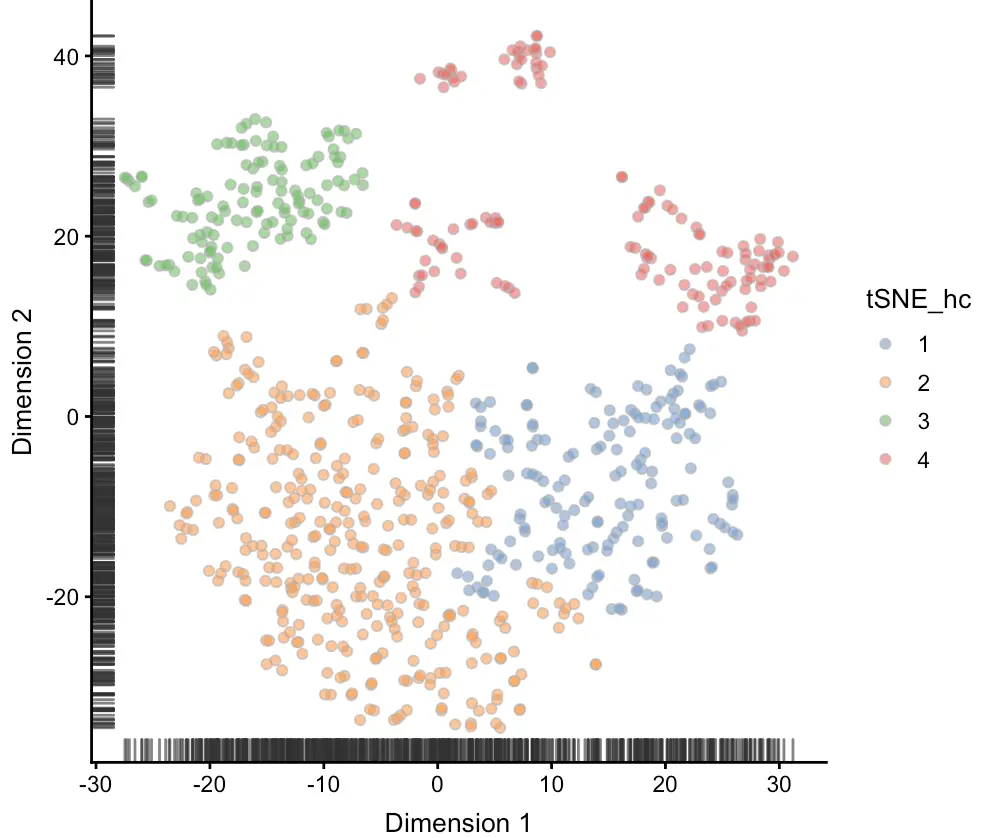
聚类3-SC3包
另外,有个R包SC3 也提供了聚类的算法,是基于K-means的,用一下试试
library(SC3) # BiocManager::install('SC3')
sce <- sc3_estimate_k(sce) # 先预估一下聚类亚群
sce@metadata$sc3$k_estimation # 预估13个亚群
rowData(sce)$feature_symbol <- rownames(rowData(sce))
# 接下来正式运行,kn参数表示的预估聚类数
# 我们这里自定义为4组
kn <- 4
start=Sys.time()
sce <- sc3(sce, ks = kn, biology = TRUE) # 运行会很慢
end=Sys.time()
(dur=end-start)
# 总共需要5分钟左右
# 会将聚类结果放入表型信息(sce@colData)中去,默认叫sc3_cluster,这里人为改个名称
sc3_cluster="sc3_4_clusters"
# 最后进行可视化--比较之前tSNE的Kmeans聚类和SC3的聚类的一致性
sc3_plot_consensus(sce, k = kn, show_pdata = c("tSNE_kmeans",sc3_cluster))
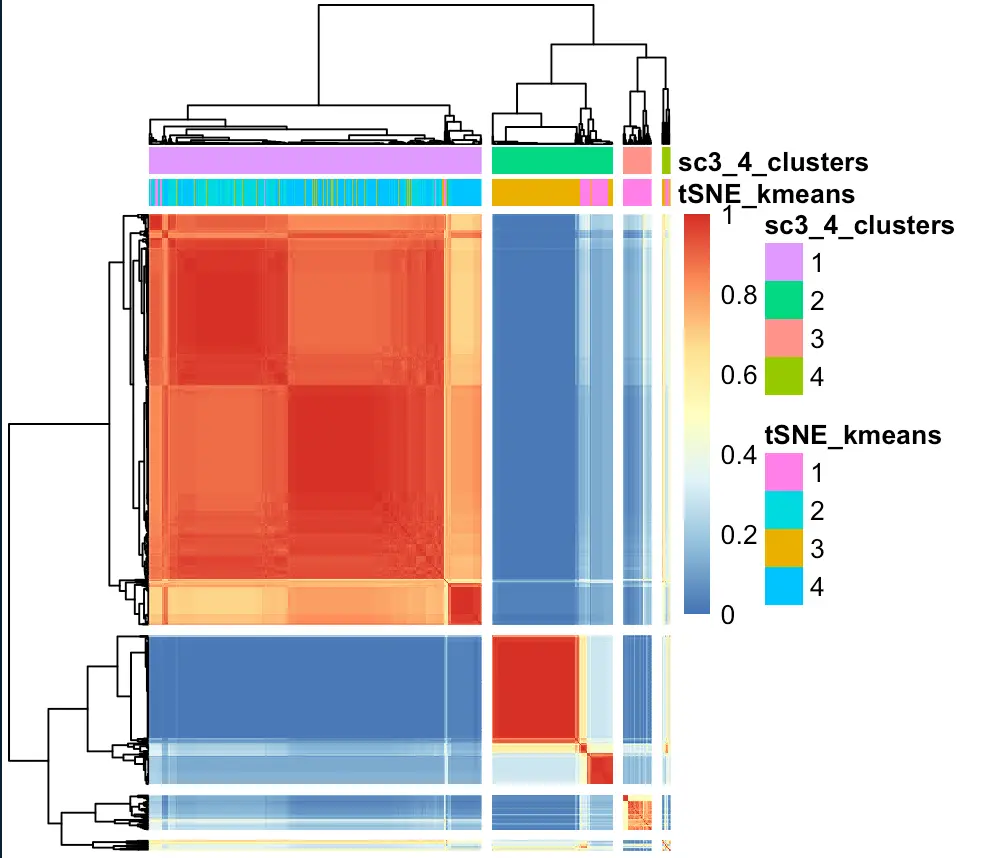
再用table函数看看二者分群的相关性:
> table(colData(sce)$tSNE_kmeans,colData(sce)$sc3_4_clusters)
1 2 3 4
1 14 42 44 6
2 194 0 0 0
3 36 147 0 7
4 278 0 0 0
同理,也能看看SC3结果与hclust的聚类相关性
sc3_plot_consensus(sce, k = kn, show_pdata = c("g",sc3_cluster))