scRNA-单细胞转录组学习笔记-12-RPKM概念及计算方法
刘小泽写于19.7.9+12 第二单元第十讲:RPKM概念及计算方法 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
RPKM须知
核心就在于基因长度的计算
参考这个网站,做的还是很清爽的:http://www.metagenomics.wiki/pdf/definition/rpkm-calculation

它的公式看上去很简单,就是对基因长度(也就是公式里的K)以及文库大小(也就是M) 进行标准化,其中文库大小很好理解,就是一个样本全部测序reads之和。但是这里有一个之前也没有注意到的问题,基因长度是什么?怎么计算出来的?
**几个问题:**它是简单的终止坐标减去起始坐标吗?它是外显子长度之和吗?那外显子之间有重合怎么处理?它是某一条转录本的长度吗?还是说基因长度是一个转录本的平均值?
这个问题是要好好总结一下,但首先还是要明确一些基因相关名词:
- 基因与转录本:一个基因可以转录成多个转录本
- 转录本与外显子:真核生物的每个转录本一般是由一个或多个外显子组成
- 转录本与cDNA:转录本逆转录得到cDNA
- 外显子与编码区CDS、非编码区UTR:可以翻译成蛋白的外显子区域是CDS区域,不能翻译的外显子开头、结尾是UTR区域
- CDS与开放阅读框ORF:ORF是从起始密码子(ATG, but not always)到终止密码子(TAA, TAG, TGA)的DNA序列,另外又有两种链的方向,因此总共有6种阅读框,它也会包含内含子(这导致了真核生物的CDS与ORF不一致;另外在原核生物中它们是一样的) http://www.scfbio-iitd.res.in/tutorial/orf.html
中心法则方向:"DNA makes RNA makes Protein”
外显子和内含子是DNA层面的gene feature,密码子(codon)是RNA层面上的feature;外显子和内含子都存在于双链DNA(dsDNA)的蛋白编码基因区域中,一般通过sense strand,也即是5’=》3‘查看
然后dsDNA变成hnRNA (不均一核RNA:heterogeneous nuclear RNA),和dsDNA的 5’-3’ 方向一致,另外将T碱基换成了U。这里虽然hnRNA中也含有dsDNA的exon、intron序列,但称呼要换:“exon transcripts"和"intron transcripts”
接着hnRNA进行了excision (‘splicing out’) 操作,切掉了intron transcripts,将剩余的exon transcripts拼接起来,得到mRNA。mRNA的序列是和DNA的exon序列一致的,它也可以视作由dsDNA的sense strand序列将T变U。密码子在DNA中叫"condons”,在mRNA中叫"triplets”
In bioinformatics, the 64 triplets are sometimes presented as a "translation table" that can be used directly with the Sense Strand sequence to infer the protein sequence.
https://www.mun.ca/biology/scarr/Exons_Introns_Codons.html
关于基因长度
既然基因包含这么多"组件”,那么求它的长度也会有几种方法:
- 选择最长的转录本
- 多个转录本的均值
- 非冗余外显子长度之和
- 非冗余CDS之和
注意到这里的**“非冗余”**,就是存在一个基因的多个外显子之间存在重叠(比如基因A的1号外显子较短,2号外显子长,1号包含在2号中),单纯的相加会重复计算
三种方法来计算基因长度
第一种:非冗余外显子之和
载入原始表达矩阵
rm(list = ls())
options(stringsAsFactors = F)
load(file = '../input.Rdata')
a[1:4,1:4]
head(df)
选择哪个包来计算基因长度呢?
不知道就搜索一下:
然后找到TxDb这个包的介绍,看Bioconductor这本电子书的41页 3.4.19节:https://bioconductor.github.io/BiocWorkshops/BioC2018.pdf
其中写道:

起步
因为处理的是小鼠数据,所以要加载相应小鼠的包:
library("TxDb.Mmusculus.UCSC.mm10.knownGene")
# 加载包以后,看看帮助文档,发现这么一句话(使用它只需要call它的名字)
## show the db object that is loaded by calling it's name
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
# 依然不知道怎么用,那么就再看帮助文档的"see also"信息
点进去发现,存在好多函数,因为我们想采用非冗余外显子加和的方法计算基因长度,因此选择exon和genes就好了
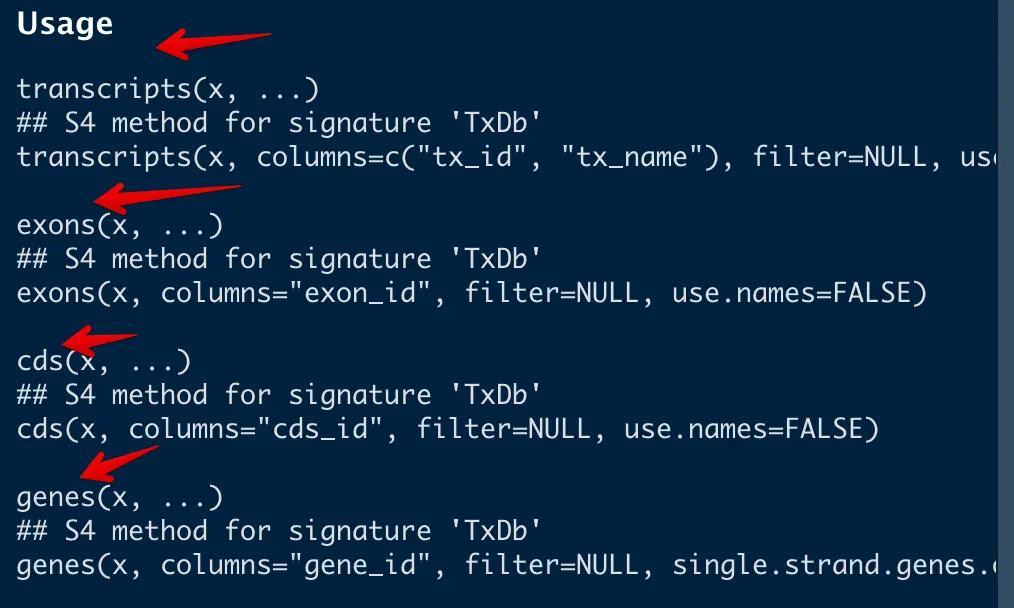
# 取出exon和gene
exon_txdb=exons(txdb)
genes_txdb=genes(txdb)
> genes_txdb
GRanges object with 24368 ranges and 1 metadata column:
seqnames ranges strand | gene_id
<Rle> <IRanges> <Rle> | <character>
100009600 chr9 21062393-21075496 - | 100009600
100009609 chr7 84940169-84964009 - | 100009609
# 看到结果是一个GRanges object,那么不懂就再搜索什么是GRanges
?GRanges
# The GRanges class is a container for the genomic locations and their associated annotations. 就是存储基因组坐标和相关注释信息的"容器"
因为取出的exon和gene都是坐标信息(可以看到上面的21062393-21075496就是坐标区间),那么二者既然都是坐标而且不一致,就会有交叉,如何找到这个交叉?
又引入了一个新的函数=》findOverlaps
o = findOverlaps(exon_txdb,genes_txdb)
> o
Hits object with 252602 hits and 0 metadata columns:
queryHits subjectHits
<integer> <integer>
[1] 1 6729
[2] 2 6729
[3] 3 6729
[4] 4 6729
[5] 5 6729
... ... ...
# 其中exon_txdb因为写在前面,于是它就作为queryHits;可以看到:它的第一个元素和genes_txdb的6729存在交叉,取出来看一下结果
> genes_txdb[6729]
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | gene_id
<Rle> <IRanges> <Rle> | <character>
18777 chr1 4807893-4846735 + | 18777
> exon_txdb[1]
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | exon_id
<Rle> <IRanges> <Rle> | <integer>
[1] chr1 4807893-4807982 + | 1
# exon的第一个元素是chr1 4807893-4807982,对应上了基因的第6729个元素:chr1 4807893-4846735,于是这个exon就属于这个基因
既然得到了所有的overlap,那就分别提取出来exon和gene的信息
t1=exon_txdb[queryHits(o)]
t2=genes_txdb[subjectHits(o)]
t1=as.data.frame(t1) #更直观地来查看t1,结果将25w个外显子的坐标提取出来
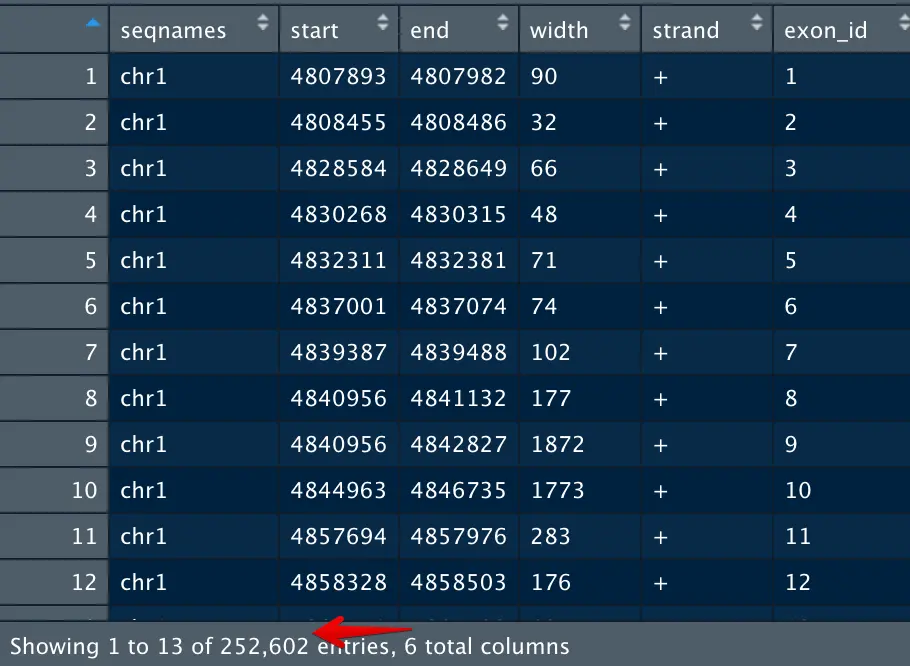
为了给t1一个对应的基因ID,需要使用t2
t1$geneid=mcols(t2)[,1]
# mcols作用是提取 a DataFrame object containing the metadata columns
# 看下面👇这个,mcols(t2)[,1]提取的也就是虚线右侧的一列"gene_id"
> genes_txdb[6729]
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | gene_id
<Rle> <IRanges> <Rle> | <character>
18777 chr1 4807893-4846735 + | 18777
再看一眼结果:
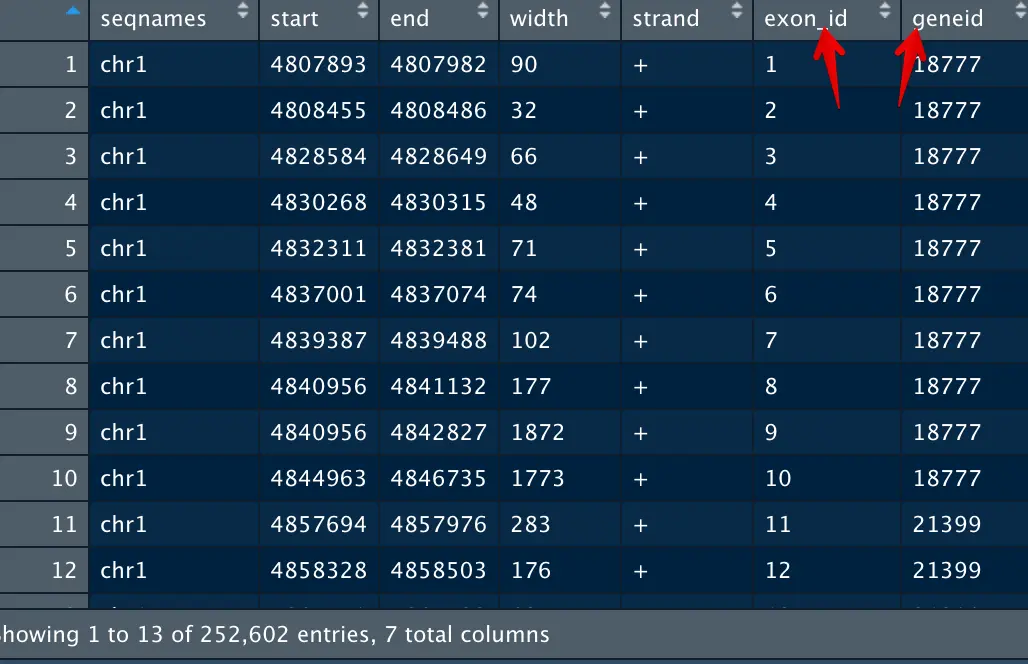
关键一步
好,看到基因18777对应着10个exon,那么如何求这个18777的长度呢?是简单把10个exon的第四列加起来吗?
**不是的!请看:第8和第9个exon,它们是不是有重叠?**或者说exon9是包含exon8的。那么应该用
sum-overlap的方法,sum好求,但overlap呢?可以这样:先不考虑具体长度值,例如现在不直接让exon1的长度为90,而是输出4807893 - 4807982这90个数值,目的就是使用
unique()函数对这些数值去重复,最后一个length就求出来了
这个思路有了,而且这个过程一定是个循环,那么第一步就是:将对应到同一个基因ID的外显子都放一起
# 利用split(x,f),需要一个x,一个f参数,其中x是向量或数据框,f是分组的因子。拆分完返回列表
split(t1,as.factor(t1$geneid))
然后第二步是对列表的每个元素取start到end的全部数值,也就是第二列到第三列,它返回的是一个列表
apply(x,1,function(y){y[2]:y[3]})
最后一步就是对列表去重、求长度。
去重有两种方法: 一是求总长度,然后去overlap;二是先去overlap,再求总长度 这里采用迂回式的第二种,也更容易操作 就是检测
y[2]:y[3],发现有重复的数字计算一遍即可,因为这一个数字就代表一个碱基位点
length(unique(unlist(tmp)))
综上,得到一个循环嵌套:
g_l = lapply(split(t1,t1$geneid),function(x){
tmp=apply(x,1,function(y){
y[2]:y[3]
})
length(unique(unlist(tmp)))
})
# 再变成一个数据框
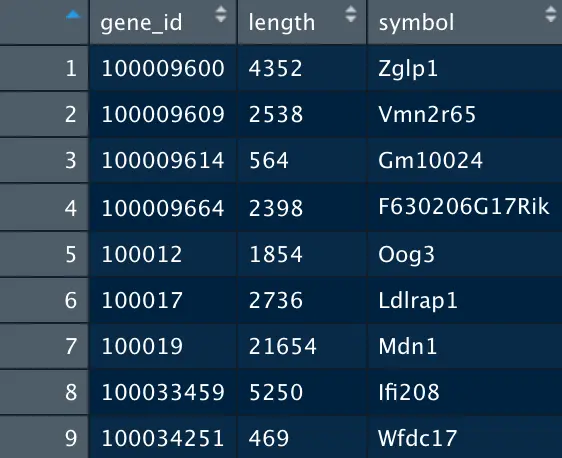
g_l=data.frame(gene_id=names(g_l),length=as.numeric(g_l))
> head(g_l)
gene_id length
1 100009600 4352
2 100009609 2538
3 100009614 564
4 100009664 2398
5 100012 1854
6 100017 2736
# 结果将gene_id对应到symbol
library(org.Mm.eg.db)
s2g=toTable(org.Mm.egSYMBOL)
g_l=merge(g_l,s2g,by='gene_id')
第二种:根据最长转录本
这个就稍微方便一点,有一个求转录本长度的函数:transcriptLengths
t_l=transcriptLengths(txdb)
> head(t_l)
tx_id tx_name gene_id nexon tx_len
1 1 uc007afg.1 18777 8 2355
2 2 uc007afh.1 18777 9 2433
3 3 uc007afi.2 21399 10 2671
4 4 uc011wht.1 21399 10 2668
5 5 uc011whu.1 21399 10 2564
6 6 uc057aty.1 <NA> 1 2719
# 发现有NA,去掉即可
t_l=na.omit(t_l)
> head(t_l)
tx_id tx_name gene_id nexon tx_len
1 1 uc007afg.1 18777 8 2355
2 2 uc007afh.1 18777 9 2433
3 3 uc007afi.2 21399 10 2671
4 4 uc011wht.1 21399 10 2668
5 5 uc011whu.1 21399 10 2564
7 7 uc007afm.2 108664 6 2396
然后看到gene_id这一列,有重复的ID,另外最后一列的length值也不同,说明这里一个基因有多个不同长度的转录本
那么就对gene_id排序(为了让同样id的基因排在一起),对tx_len排序(为了找最长的转录本)
t_l=t_l[order(t_l$gene_id,t_l$tx_len,decreasing = T),]
> head(t_l)
tx_id tx_name gene_id nexon tx_len
15232 15232 uc008vih.2 99982 20 3075
15231 15231 uc008vig.2 99982 19 3015
9376 9376 uc008pkr.1 99929 6 4154
11784 11784 uc008rsk.3 99899 8 2909
11479 11479 uc008rat.3 99890 2 3065
11480 11480 uc008rau.2 99890 1 2475
# 上面👆可以看到,99982这个基因就会选取第一个转录本15232,因为它的长度为3075
# 其实这样降序排列完,就可以直接去重,保留最长的那一个(就是第一个)了
t_l=t_l[!duplicated(t_l$gene_id),]
> head(t_l)
tx_id tx_name gene_id nexon tx_len
15232 15232 uc008vih.2 99982 20 3075
9376 9376 uc008pkr.1 99929 6 4154
11784 11784 uc008rsk.3 99899 8 2909
11479 11479 uc008rat.3 99890 2 3065
10866 10866 uc008pqg.3 99889 10 3248
11552 11552 uc008rdv.2 99887 8 6271
有了基因长度,计算RPKM
上面得到了有长度信息的基因,首先要和原始矩阵的行名取交集
ng=intersect(rownames(a),g_l$symbol)
# 得到的新表达矩阵=》有长度信息的基因表达矩阵
exprSet=a[ng,]
lengths=g_l[match(ng,g_l$symbol),2] #
> head(lengths)
[1] 1122 795 619 4556 1743 1471
> head(rownames(exprSet))
[1] "0610005C13Rik" "0610009B22Rik" "0610009L18Rik" "0610010F05Rik"
[5] "0610010K14Rik" "0610012G03Rik"
# 简单探索
> exprSet[1:3,1:3]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5
0610005C13Rik 0 0 0
0610009B22Rik 0 0 0
0610009L18Rik 0 0 0
> dim(exprSet)
[1] 22731 768
接下来就是对新表达矩阵进行操作:
先求文库大小:
total_count<- colSums(exprSet)
> head(total_count)
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5 SS2_15_0048_A4
95099 92537 161260 121297
SS2_15_0048_A1 SS2_15_0048_A2
263927 280547
然后需要对每个基因表达量值除以对应的基因长度(单位是Kb),再除以总文库(单位是Mb)大小
# 如果说i代表一行表达量,exprSet[,i]就是表达矩阵的第一列,即22731个基因的表达量,lengths就是22731个基因的长度,它和exprSet[,i]是一一对应的。total_count[i]就是第一个样本的文库大小。最后需要乘以10^9来抵消单位的影响
10^9*exprSet[,i]/lengths/total_count[i]
再做一个循环:
实现了从第一个样本到最后一个求得RPKM,结果返回一个包含768个元素的列表,每个样本求得的RPKM值占一行
lapply(1:length(total_count),
function(i){
10^9*exprSet[,i]/lengths/total_count[i]
})
接着对每个元素进行rbind操作,拼接到一起,但是拼接的结果是:768行,22731列
do.call(rbind,
lapply(1:length(total_count),
function(i){
10^9*exprSet[,i]/lengths/total_count[i]
}))
最后要转置一下:
rpkm <- t(do.call( rbind,
lapply(1:length(total_count),
function(i){
10^9*exprSet[,i]/lengths/total_count[i]
}) ))
# 检查下
> rpkm[1:4,1:4]
[,1] [,2] [,3] [,4]
[1,] 0 0 0 7.347796
[2,] 0 0 0 0.000000
[3,] 0 0 0 0.000000
[4,] 0 0 0 19.904850
回顾总结
下面就以刚刚计算出来的第一行,第四列的
7.347796这个值为例,看看到底RPKM是怎么算出来的,算是一个复习
首先看下表达矩阵:
> exprSet[1:4,1:4]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5
0610005C13Rik 0 0 0
0610009B22Rik 0 0 0
0610009L18Rik 0 0 0
0610010F05Rik 0 0 0
SS2_15_0048_A4
0610005C13Rik 1
0610009B22Rik 0
0610009L18Rik 0
0610010F05Rik 11
# 第一行第四列,也就是0610005C13Rik基因在SS2_15_0048_A4样本中的原始表达量为1
# 然后总的文库大小是121297
> total_count[4]
SS2_15_0048_A4
121297
# 它的第一个基因0610005C13Rik长度是1122
> lengths[1]
[1] 1122
# 因此计算公式就是
# 10^9*exprSet[,i]/lengths/total_count[i]
> 10^9*1/1122/121297
[1] 7.347796
从这个计算结果也可以看到:RPKM值为7,它的count值才为1,这样会过滤掉很多存在RPKM表达量的基因,因此过滤基因设定count值为0就好
注意:不要认为count值为1了,RPKM就是7左右。这个要取决于文库大小,如果文库很小,那么count为1时,RPKM也能达到几万
最后和文章提供的RPKM矩阵比较下:
a=read.table('../GSE111229_Mammary_Tumor_fibroblasts_768samples_rpkmNormalized.txt.gz',
header = T ,sep = '\t')
a[1:4,1:4]
rpkm_paper=a[ng,]
# 文章做的
> rpkm_paper[1:4,1:4]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5
0610005C13Rik 0 0 0
0610009B22Rik 0 0 0
0610009L18Rik 0 0 0
0610010F05Rik 0 0 0
SS2_15_0048_A4
0610005C13Rik 6.966712
0610009B22Rik 0.000000
0610009L18Rik 0.000000
0610010F05Rik 19.843349
# 我们做的
> rpkm[1:4,1:4]
[,1] [,2] [,3] [,4]
[1,] 0 0 0 7.347796
[2,] 0 0 0 0.000000
[3,] 0 0 0 0.000000
[4,] 0 0 0 19.904850
可以看到有一点差别,但差别不大,这是因为计算的基因长度方法是有差别的