scRNA-单细胞转录组学习笔记-7
刘小泽写于19.7.4 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53 第二单元第五讲:重复平均表达量和变异系数相关性散点图
前言
这一次的目的是重复文章附件中的一张图:
附件地址在:https://static-content.springer.com/esm/art%3A10.1038%2Fs41467-018-07582-3/MediaObjects/41467_2018_7582_MOESM1_ESM.pdf
存在这样一张图:
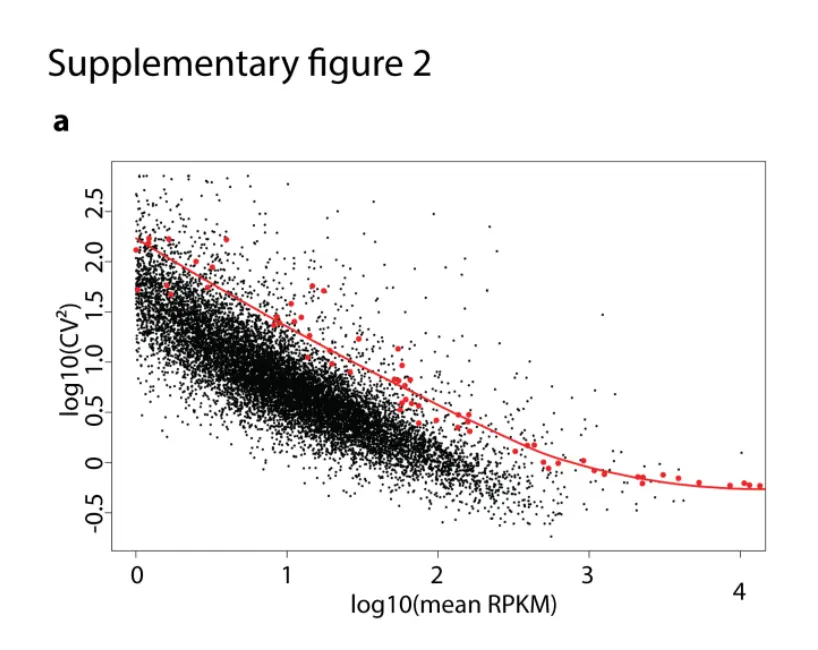
图片分析
首先看横坐标,不论是RPKM还是原始count都是表达值,然后做了均值的log10处理;然后纵坐标是CV值,它表示变异系数(coefficient of variation),也是先求CV的平方,然后做log10处理
来看一下什么是CV值?
变异系数又称离散系数或相对偏差(我们肯定都听过标准偏差,也就是sd值,它描述了数据值偏离算术平均值的程度),这个相对偏差描述的是标准偏差与平均值之比,即:cv=sd/mean*100% 。
为何不用sd而用cv值呢?
先说说sd值,它和均值mean、方差var一样,都是对一维数据进行的分析,需要数据满足两个条件:中部、单峰。也就是说数据集只存在一个峰值,并且这个峰值大致位于数据集的中部。另外当比较两组数据集的离散程度大小时,即使它们各自满足"中部单峰"的条件,如果出现两组数据测量尺度差别太大或数据量纲存在差异的话,直接用标准差就不合适了
变异系数就可以解决这个问题,它利用原始数据标准差和原始数据平均值的比值来各自消除尺度与量纲的差异。
CV的意义
https://www.biostars.org/p/272801/
Genes which are stably expressed across replicates/experiments as the CV is low (0.5).
代码计算
第一步:清空变量,加载数据
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
load(file = '../input_rpkm.Rdata')
# 就会得到RPKM的过滤表达矩阵dat和细胞信息metadata(包括clust分群、细胞板信息、检测到的基因数量)
> head(metadata)
g plate n_g all
SS2_15_0048_A3 1 0048 3065 all
SS2_15_0048_A6 2 0048 3036 all
SS2_15_0048_A5 1 0048 3742 all
SS2_15_0048_A4 3 0048 5012 all
SS2_15_0048_A1 1 0048 5126 all
SS2_15_0048_A2 3 0048 5692 all
第二步:计算mean、sd值
mean_per_gene <- apply(dat, 1, mean, na.rm = TRUE) #对表达矩阵每行求均值
sd_per_gene <- apply(dat, 1, sd, na.rm = TRUE) #同理求标准差
第三步:构建数据框,计算cv值
cv_per_gene <- data.frame(mean = mean_per_gene,
sd = sd_per_gene,
cv = sd_per_gene/mean_per_gene)
# 然后赋予行名
rownames(cv_per_gene) <- rownames(dat)
> head(cv_per_gene)
mean sd cv
0610007P14Rik 25.634318 48.60264 1.895999
0610009B22Rik 27.203266 64.41918 2.368068
0610009L18Rik 3.864324 19.61355 5.075544
0610009O20Rik 10.808251 26.01667 2.407112
0610010F05Rik 8.194137 21.06394 2.570612
0610010K14Rik 31.812982 52.29101 1.643701
作图,一点点优化
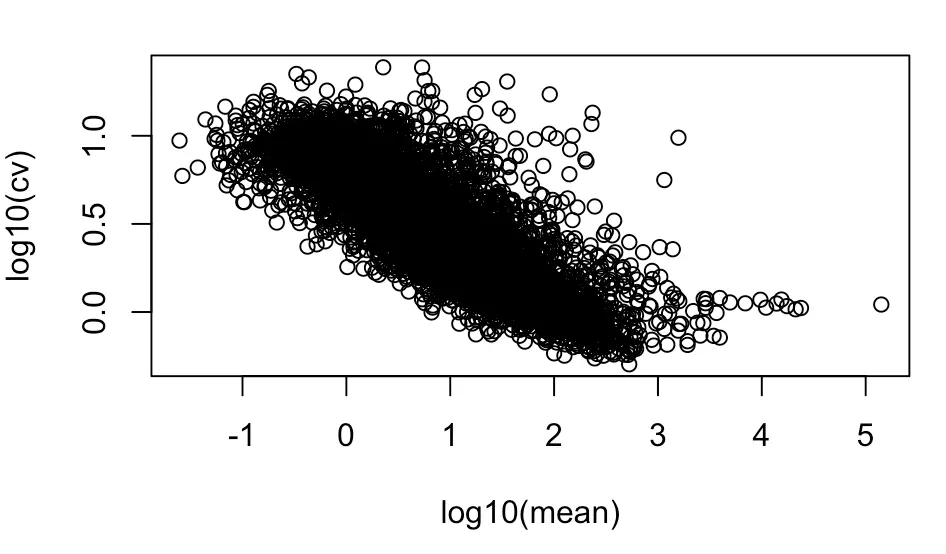
先画第一张图—纵坐标为cv
with(cv_per_gene,plot(log10(mean),log10(cv)))
# with实现的功能就是将逗号后面的操作对象限定在逗号前面的数据框中,就不用每次都引用数据框了
然后画第二张图—纵坐标为cv平方
with(cv_per_gene,plot(log10(mean),log10(cv^2)))
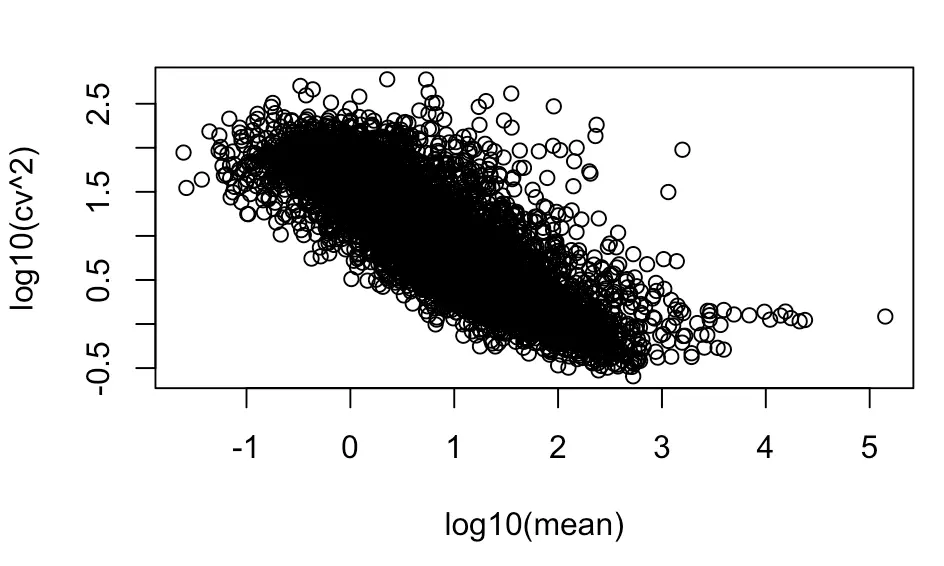
发现纵坐标的区间发生改变,点的位置没有改变,好了,初见雏形,和原图最大的差别是趋势线
然后画第三张图—添加趋势线
为了更方便地模拟原始数据,先在CV的数据框中添加两列:log10cv2和log10mean;另外看到我们上面作的图中x轴范围是-1~5,而文章的图中是0~4
cv_per_gene$log10cv2=log10(cv_per_gene$cv^2)
cv_per_gene$log10mean=log10(cv_per_gene$mean)
# 修改x轴坐标范围为0-4
cv_per_gene=cv_per_gene[cv_per_gene$log10mean < 4, ]
cv_per_gene=cv_per_gene[cv_per_gene$log10mean > 0, ]
library(ggpubr)
ggscatter(cv_per_gene, x = 'log10mean', y = 'log10cv2',
color = "black", shape = 16, size = 1, # Points color, shape and size
xlab = 'log10(mean RPKM)', ylab = "log10(cv^2)",
add = "loess", #添加拟合曲线
add.params = list(color = "red",fill = "lightgray"),
cor.coeff.args = list(method = "spearman"),
label.x = 3,label.sep = "\n",
dot.size = 2,
ylim=c(-0.5, 2.5),
xlim=c(0,4)
)
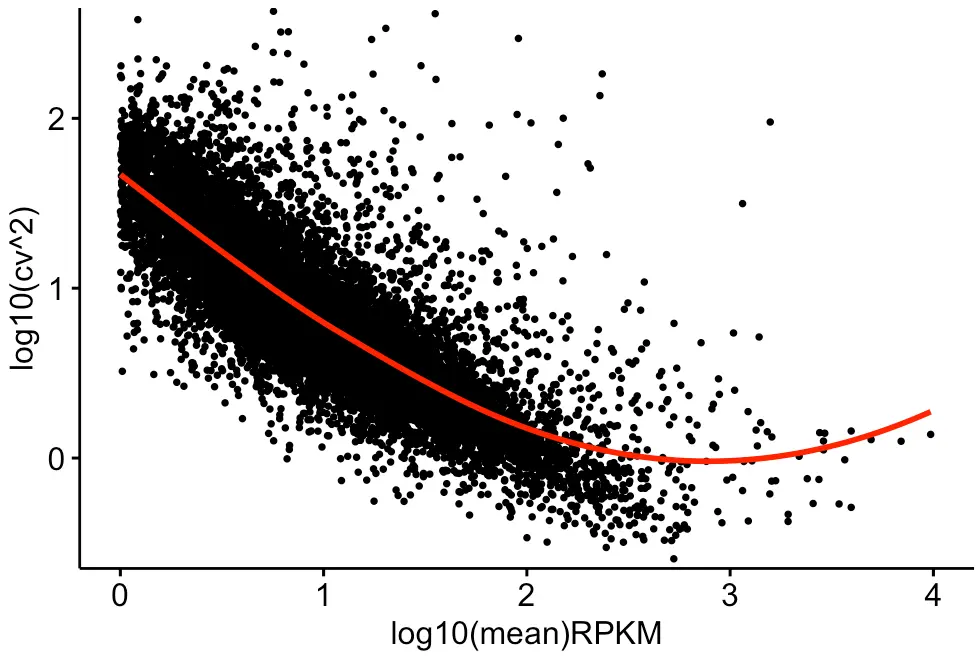
发现和原文的差别还是蛮大的,为什么呢?趋势线添加
loess不应该有这么明显的上升趋势才对,想到原文的注释可能是最好的答案,又返回看了原文,发现这么一句话
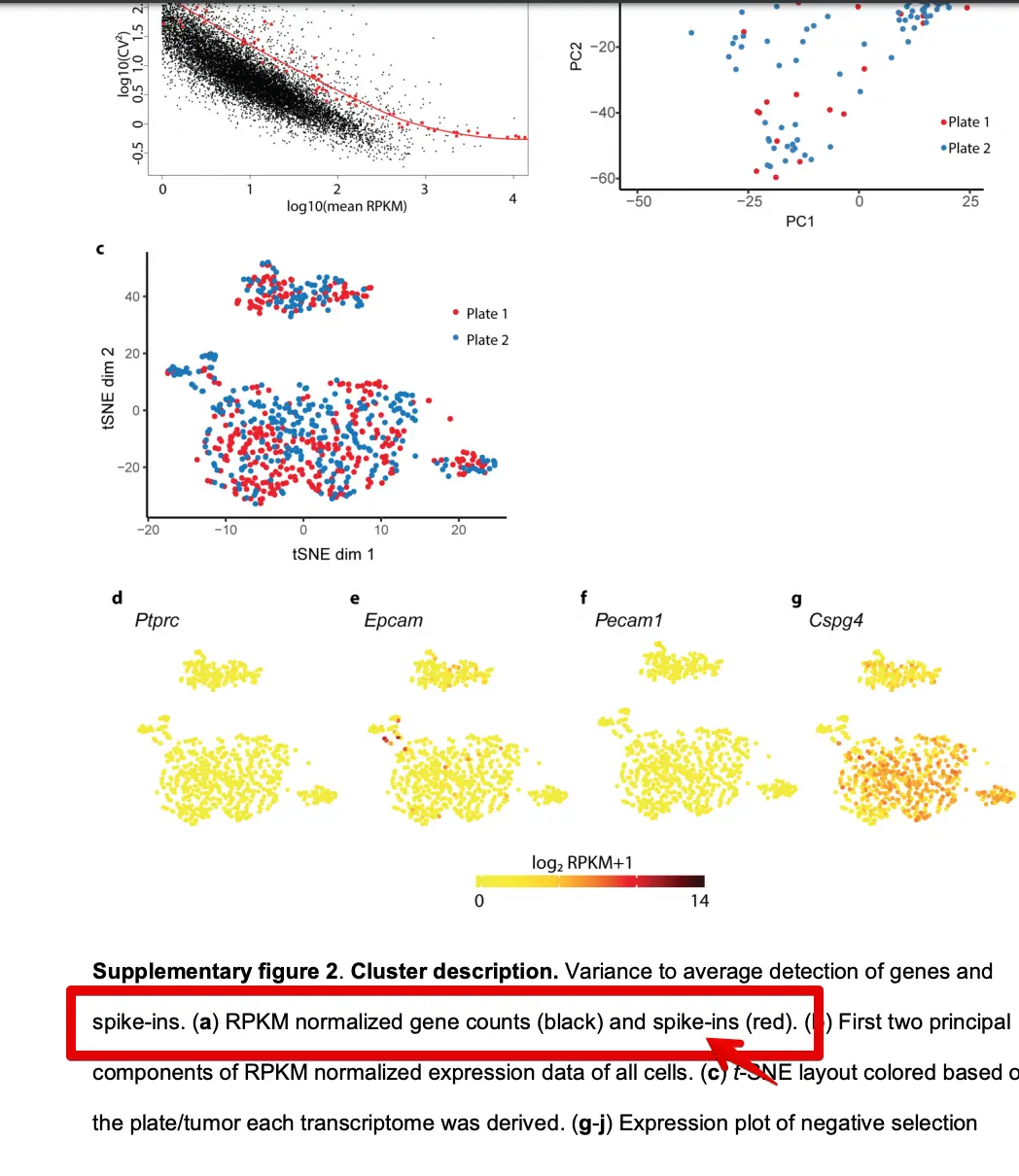
原来趋势线不是指全部的基因,而是ERCC spike-in
> grep("ERCC",rownames(cv_per_gene))
[1] 11475 11476 11477 11478 11479 11480 11481 11482 11483 11484 11485
[12] 11486 11487 11488 11489 11490 11491 11492 11493 11494 11495 11496
[23] 11497 11498 11499 11500 11501 11502 11503 11504 11505 11506 11507
[34] 11508 11509 11510 11511 11512 11513 11514 11515 11516 11517 11518
[45] 11519 11520 11521 11522 11523 11524 11525 11526 11527 11528 11529
[56] 11530
> tail(rownames(cv_per_gene))
[1] "ERCC-00160" "ERCC-00162" "ERCC-00163" "ERCC-00165" "ERCC-00170"
[6] "ERCC-00171"
于是需要做两个图层,其中一个需要挑出来ERCC,然后单独做,然后搜索了ggscatter的帮助信息,发现只需要增加一个ggp参数就可以做到
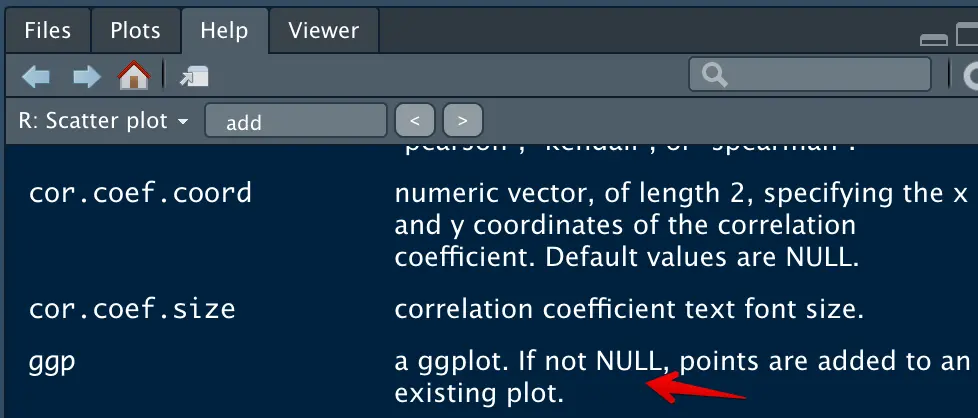
ggscatter(cv_per_gene[-grep("ERCC",rownames(cv_per_gene)),], x = 'log10mean', y = 'log10cv2',
color = "black", shape = 16, size = 1, # Points color, shape and size
xlab = 'log10(mean RPKM)', ylab = "log10(CV^2)",
mean.point=T,
cor.coeff.args = list(method = "spearman"),
label.x = 3,label.sep = "\n",
dot.size = 2,
ylim=c(-0.5, 2.5),
xlim=c(0,4),
# 这里ggp参数的意思就是:增加一个ggplot图层
ggp = ggscatter(cv_per_gene[grep("ERCC",rownames(cv_per_gene)),], x = 'log10mean', y = 'log10cv2',
color = "red", shape = 16, size = 1, # Points color, shape and size
xlab = 'log10(mean RPKM)', ylab = "log10(CV^2)",
add = "loess", #添加拟合曲线
mean.point=T,
add.params = list(color = "red",fill = "lightgray"),
cor.coeff.args = list(method = "pearson"),
label.x = 3,label.sep = "\n",
dot.size = 2,
)
)
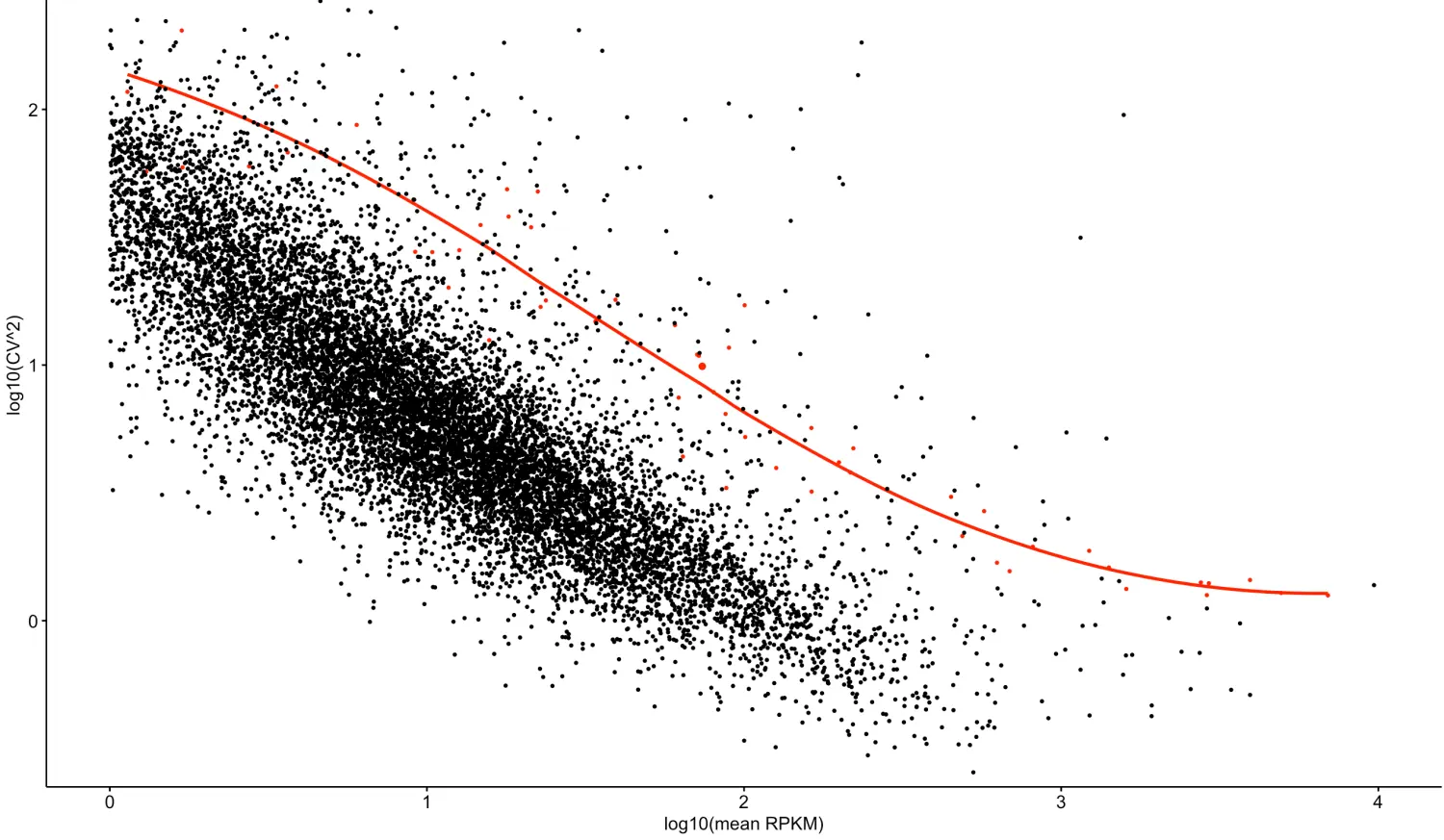
写写自己的理解
再看原文,发现这个附图2.a主要是用来说明ERCC的,也就是做了一个技术误差检测,变异系数对两个或多个数据集进行比较时,如果度量单位保持平均数相同,那么可以直接比较标准差,也就是说,设定同一个横坐标(比如1),然后红线是ERCC,衡量技术误差,如果我们测得基因在ERCC以下,说明我们测得基因sd值小于ERCC标准的,说明基因的技术误差也是在可接受范围之内