scRNA-单细胞转录组学习笔记-3
刘小泽写于19.6.13 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53 第二单元第一讲:单细胞转录组上游分析之shell回顾
前言
目前主流的单细胞测序技术主要有两种:主打基因数量的smart-seq2和主打细胞数量的10X Genomics。单细胞转录组分析和常规的转录组分析没有太大区别,只是将原来作为一个样本的一块组织给分解,变成大量细胞,并且每个细胞单独作为一个样本;就像TCGA这样的大型人群队列中测一千个人的转录组,只不过一次性将这一千个人的转录组放在一起进行分析。
单细胞数据的一个特点就是:每个样本的数据量小。以人为例,常规转录组一般都能测30M、50M,也就是动辄几千万条reads。但是单细胞能够测500万条reads就非常厉害了
单细胞上游分析需要get的点
需要用到linux、R以及常规转录组分析流程
拿数据
首先拿到文章,先搜索"GEO"或者"GSE”

然后点进去超链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111229,就到了GEO数据库,注意三点就可以:
只需要改链接最后的
GSExxxxx就可以快速访问不同的GEO数据要是想下载作者做好的表达矩阵,然后直接进行下游分析

要是想从原始测序数据fq文件开始自己分析,就要进入SRA测序数据中心

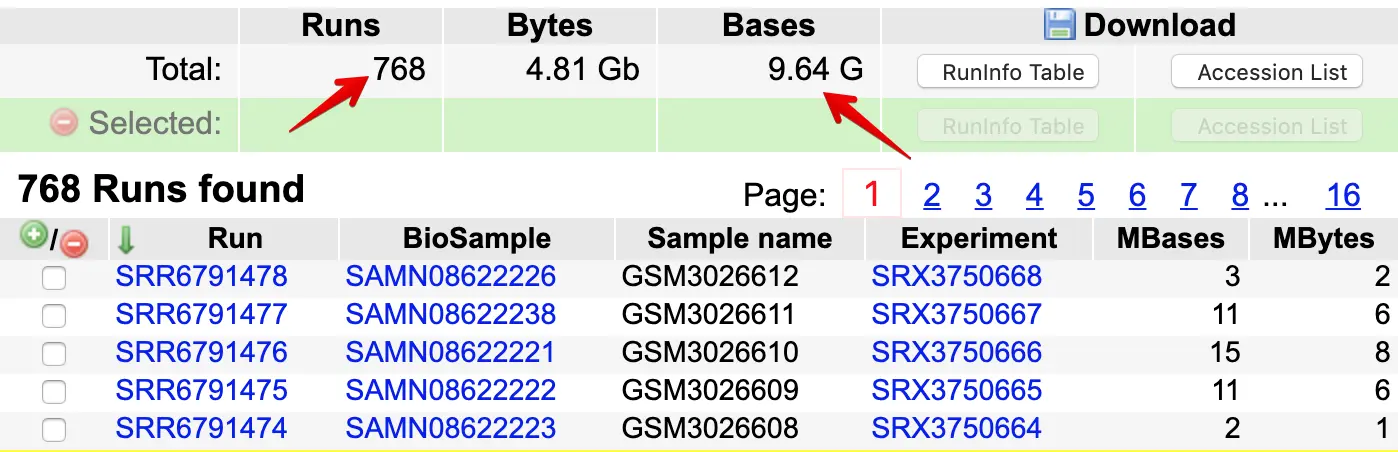
进入测序数据中心就可以看到:接近800个细胞才测了不到10G的数据量,要知道一个常规转录组数据都可以做到10G数据量 (另外,10X的数据量要比smart-seq2还要小)。
下载+转换
利用conda安装sra-tools 就会带prefetch ,然后利用这个软件下载sra原始数据,默认使用http方式下载,如果网速比较慢,还可以使用"开挂"模式,具体操作看:
来吧,加速你的下载
下载全部的sra文件后会发现:smart-seq2结果中每个细胞都是一个单独的sra文件,它是单细胞的单样本
这一点和10X是有区别的,10X是一个样本中就包含了4000-8000个细胞,但不会拆分成4000-8000个fq文件,需要进一步利用UMI、barcode将细胞分开,也就是说,10X多了一步拆分的过程。

有了sra,就需要转换成fastq,利用的软件就是fastq-dump ,它也属于sra-tools ,一般参数设定为:fastq-dump --gzip --split-3 -O ./
因为采用了3’单端测序,因此转换后也只有一个fq文件(如果是采用10X,虽然也是单端测序,但是它转换结果会是三个文件:sample index、barcode+UMI、真正的测序reads)
拿到原始数据后一般要利用fastqc和multiqc进行质控,质量合格进入下一环节
比对
利用hisat2 ,需要注意:先利用hisat2-build命令构建基因组索引,文章使用的是小鼠的基因组mm10
只要有一台服务器存在这个索引即可,可以使用跨服务器拷贝
scp命令 使用方法简单: https://linuxtools-rst.readthedocs.io/zh_CN/latest/tool/scp.htmlscp local_file remote_username@remote_ip:remote_folder # 拷贝整个目录就加参数-r
# 因为是单端测序文件,因此hisat2软件就要用-U选项(如果双端的话,直接用 -1 -2 选项即可)
index=/PATH_to_hisat2_mm10/
ls *.gz | while read i;do hisat2 -p 10 -x $index -U $i -S ${i%%.*}.hisat.sam;done
# -S选项指的是输入比对结果SAM文件,它的参数就是输出的SAM叫什么。看到其中有一个%%.*,它的意思就是取我们输入原始测序文件(i)的前缀
直接运行的话,会给出比对结果,看一下比对率
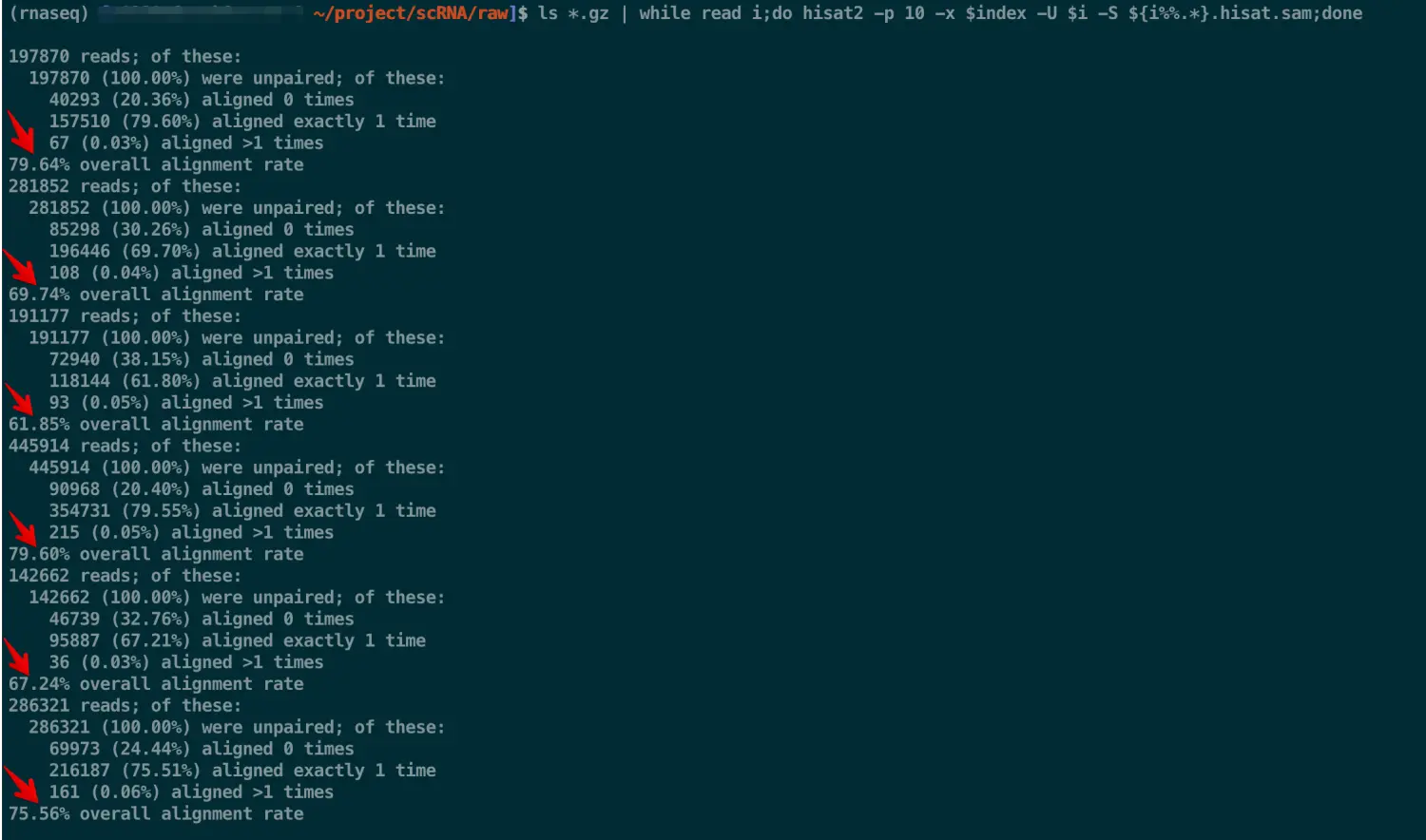
得到的结果:
比对后SAM要转为BAM才能进行下一步定量,利用samtools
ls *.sam|while read i ;do (samtools sort -O bam -@ 10 -o $(basename ${i} ".sam").bam ${i});done
# 其中一个小tip就是:basename这里,会返回每个$i,也就是每个sam文件的名称,然后".sam"就是将名称中的.sam去掉,于是只留下了前缀名,这样才能更改文件名为bam
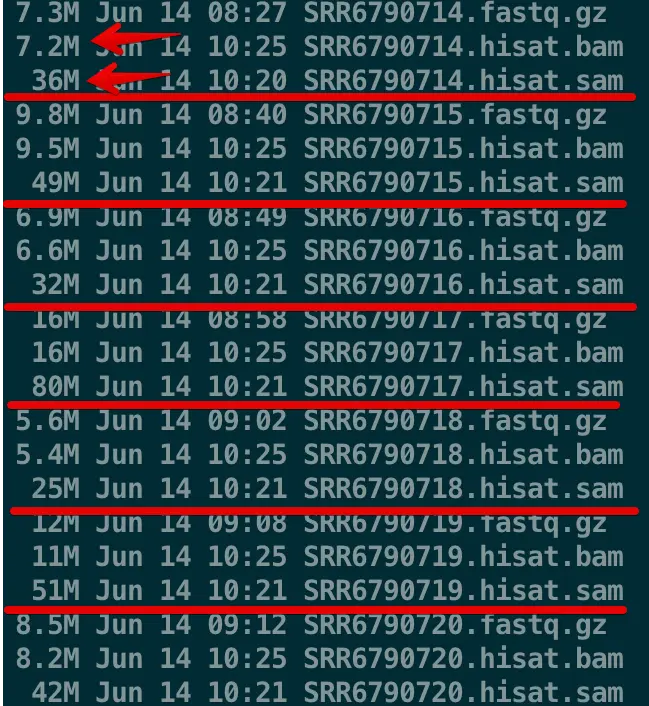
可以看到bam比sam小了很多倍
转换后继续构建索引:
ls *.bam |xargs -i samtools index {}
定量
它的目的就是将bam比对结果和参考的基因注释(其实也是基因在基因组上的位置信息)进行比较,看看我们的结果中对应了哪些基因。主要利用featureCounts
GTF文件可以去Gencode数据库下载: ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M21/gencode.vM21.annotation.gtf.gz
# 还是先指定gtf的位置
gtf=/YOUR_GTF_PATH/
featureCounts -T 5 -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
# 如果是双端测序,可以加上-p选项
关于这款软件: featureCounts官网
featureCounts不仅支持基因或转录本的定量,还可以进行exons, promoter, gene bodies, genomic bins and chromosomal locations的定量
这个软件不是单独下载的,它是集成在subread软件中,因此只要下载好subread就能使用featureCounts。
它需要的输入文件也很简单:比对的sam/bam文件(我们经常使用bam,是因为它占据硬盘空间小)、注释文件GTF
它的定量有两个层次:一个是对
feature定量,另一个是对metafeature进行定量。官网对它们定义的描述:Each entry in the provided annotation file is taken as a feature (e.g. an exon). A meta-feature is the aggregation of a set of features (e.g. a gene).
feature也就是基因组区间最小的信息(如外显子);metafeature可以是多个feature的组合,如同一个基因的多个外显子集合;因此这款软件可以单独对外显子定量,也可以对基因进行定量。但只有比对到多个不同区间时,才会分别计数
-T表示线程数,默认是1;-t表示要计数的feature名称,也就是GTF的第三列信息,默认是exon;-g表示提取的GTF最后一列attribute信息,默认是gene_id;-a指定使用的注释文件;-o是输出文件
得到的结果中可以看到,基因对应的外显子、起始终止坐标等。其中大量的基因在7个示例样本中都没有表达量,也就是说,对于大部分细胞来说许多基因都是测不到的

得到的表达矩阵也就是文中一开始提到的作者给出的rawcounts表达矩阵,只不过这里我们只有7个样本,而真正有768个样本。另外表达矩阵的软件、版本、参数有所差别,因此得到的不会完全一样,这也是可接受的