scRNA-使用sctransform去除批次效应
刘小泽写于19.10.10 上一次介绍了 使用Seurat 的merge函数来合并4个样本(各有2个生物学重复)的8组数据,但是merge只是将原始数据简单混合起来,谁也不知道混合后的结果是不是引入了批次效应
关于去除多组数据中的批次效应,有多种算法,如Seurat包的CCA(canonical correlation analysis)、LIGER的NMF(non-negative matrix factorization)、 Scran包的mnnCorrect、Seurat包的sctransform
这次就来看看sctransform是如何使用的
前言
目前教程更新于2019-10-08
https://satijalab.org/seurat/v3.1/sctransform_vignette.html
它的开发者曾说:

还支持管道单行命令:
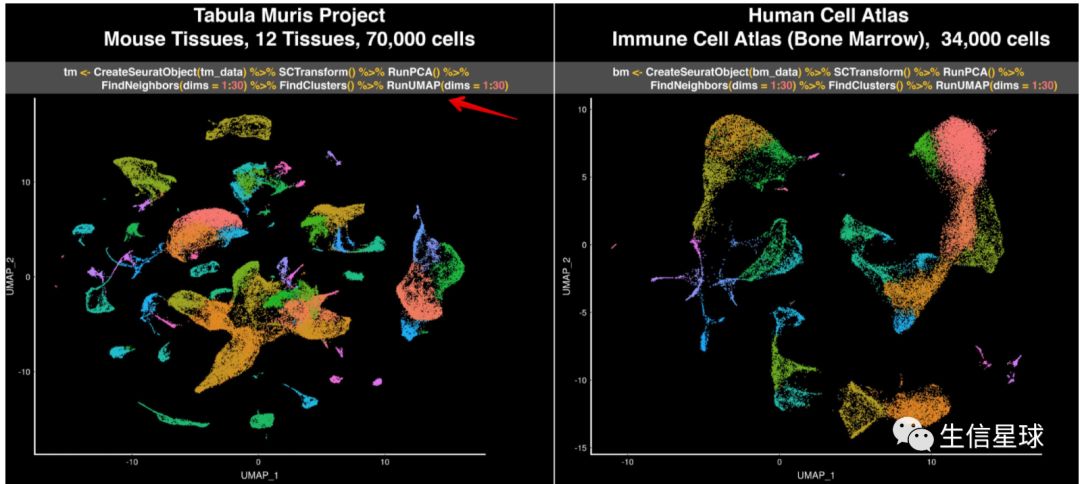
之前利用常规流程处理,得到的UMAP结果是:

现在再使用sctransform看看结果
第一步:加载10X原始数据,创建对象
# 原始数据在:https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
pbmc_data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc_data)
第二步:记录线粒体信息,一会进行校正
pbmc <- PercentageFeatureSet(pbmc, pattern = "^MT-", col.name = "percent.mt")
pbmc <- SCTransform(pbmc, vars.to.regress = "percent.mt", verbose = FALSE)
别看SCTransform只有一个单独的函数,其实它做了:
NormalizeData、ScaleData、FindVariableFeatures的事情,并且也支持ScaleData的vars.to.regress运行的结果存储在:
pbmc@assays$SCT)或者pbmc[["SCT"]]> dim(pbmc@assays$RNA) [1] 32738 2700 > dim(pbmc@assays$SCT) [1] 12572 2700 > pbmc@assays$SCT Assay data with 12572 features for 2700 cells Top 10 variable features: S100A9, GNLY, LYZ, S100A8, NKG7, FTL, GZMB, IGLL5, CCL5, FTH1
第三步:PCA+UMAP降维,然后聚类
pbmc <- RunPCA(pbmc, verbose = FALSE)
pbmc <- RunUMAP(pbmc, dims = 1:30, verbose = FALSE)
pbmc <- FindNeighbors(pbmc, dims = 1:30, verbose = FALSE)
pbmc <- FindClusters(pbmc, verbose = FALSE)
DimPlot(pbmc, label = TRUE) + NoLegend()
得到的结果是:
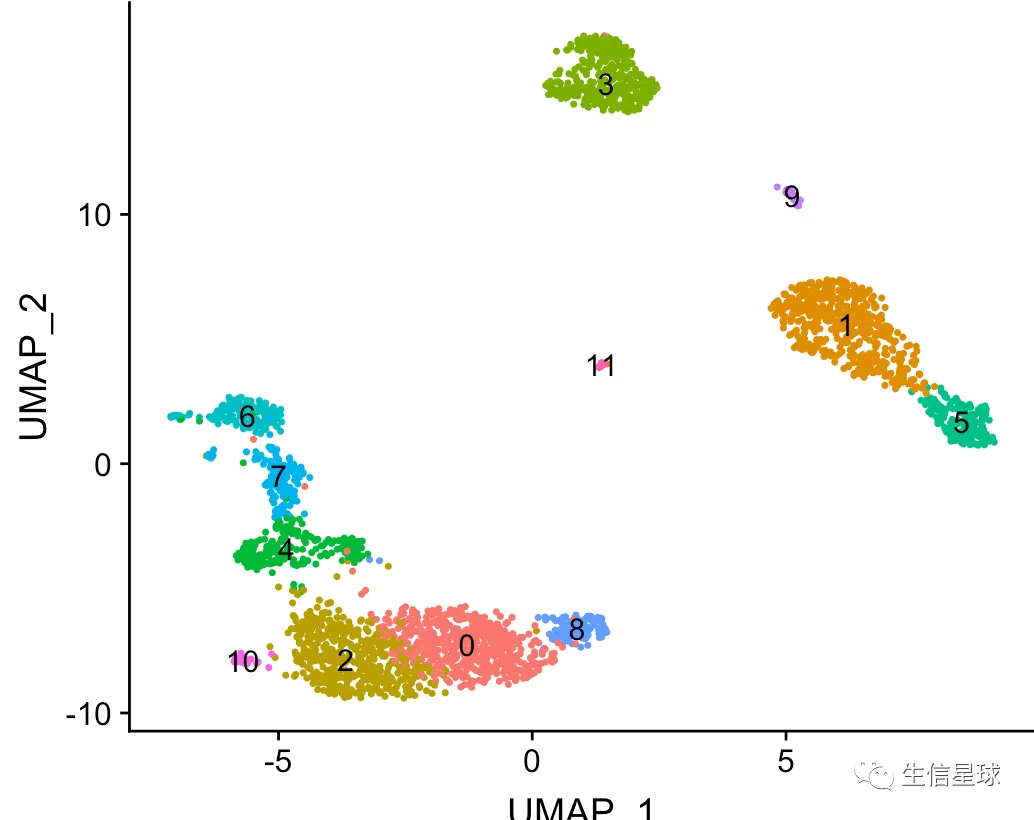
注意到:这里的FindNeighbors使用了更多的主成分(30个),而之前常规分析中根据ElbowPlot(pbmc)结果仅使用了10个主成分就得了不错的结果。
这是因为:
- 在常规分析中,使用少量的PC既能关注到关键的生物学差异,又能够不引入更多的技术差异,相当于一种保守性的做法。是的,它会失去一些生物差异信息,但是同时又在常规手段中比较安全。
- 这里使用的
sctransform,显然更“自信”一些,它认为:我很厉害,我的归一化、标准化都做得不错,多给我一些PCs吧,我能提取更多的生物差异,并且兼顾不引入技术误差
另外,常规分析中的FindVariableFeatures默认得到2000个高变异基因(HVGs),而这里的sctransform因为使用了更多的PCs,算法也更优化,所以默认会得到3000个HVGs。sctransform认为:新增加的这1000个基因就包含了之前没有检测到的微弱的生物学差异。而且,即使使用全部的全部的基因去做下游分析,得到的结果也是和sctransform这3000个基因的结果相似
综合:单行代码实现分析
因为SCTransform对参数的要求不是很多,一般默认参数就能应付大多数情况,因此作者也给出了单行从创建对象到最后分群的结果
pbmc <- CreateSeuratObject(pbmc_data) %>% PercentageFeatureSet(pattern = "^MT-", col.name = "percent.mt") %>%
SCTransform(vars.to.regress = "percent.mt") %>% RunPCA() %>% FindNeighbors(dims = 1:30) %>%
RunUMAP(dims = 1:30) %>% FindClusters()
关于SCTransform得到的结果
作为了解即可:它利用了正则化负二项分布(regularized negative binomial regression)计算了技术噪音模型,得到的残差是归一化值,有正有负。正值表示:考虑到细胞群体中基因的平均表达量和细胞测序深度,某个细胞的某个基因所包含的UMIs比预测值要高。
它的结果有以下几种,不过都包含在pbmc[["SCT"]]
pbmc[["SCT"]]@scale.data:包含了残差数据,用作PCA的输入。这个数据不是稀疏矩阵,因此会占用大量内存。不过SCTransform函数计算的时候,为了节省内存,默认使用了return.only.var.genes = TRUE,只保留差异基因的结果pbmc[["SCT"]]@counts:包含了校正后的UMI count值pbmc[["SCT"]]@data:包含了上面count值的log-normalized结果,有利于后面可视化- 目前可以使用
pbmc[["SCT"]]@data结果进行差异分析,但实际上,官方更推荐直接使用残差值pbmc[["SCT"]]@scale.data【这个功能目前还不支持,会在后面的Seurat版本中更新】
第四步:使用一些权威的marker对细胞群体注释
这些marker的选择:
- CD8 T cell populations (naive, memory, effector), based on CD8A, GZMK, CCL5, GZMK expression
- CD4 T cell populations (naive, memory, IFN-activated) based on S100A4, CCR7, IL32, and ISG15
- Additional developmental sub-structure in B cell cluster, based on TCL1A, FCER2
- Additional separation of NK cells into CD56dim vs. bright clusters, based on XCL1 and FCGR3A
VlnPlot(pbmc, features = c("CD8A", "GZMK", "CCL5", "S100A4", "ANXA1", "CCR7", "ISG15", "CD3D"),
pt.size = 0.2, ncol = 4)
FeaturePlot(pbmc, features = c("CD8A", "GZMK", "CCL5", "S100A4", "ANXA1", "CCR7"), pt.size = 0.2,
ncol = 3)
FeaturePlot(pbmc, features = c("CD3D", "ISG15", "TCL1A", "FCER2", "XCL1", "FCGR3A"), pt.size = 0.2,
ncol = 3)