scRNA-组合多个单细胞转录组数据
刘小泽写于19.10.8 前几天单细胞天地推送了一篇整合scRNA数据的文章: 使用seurat3的merge功能整合8个10X单细胞转录组样本 这次根据推送,再结合自己的理解写一写
前言
单细胞数据未来会朝着多样本发展,因此数据整合是一项必备技能。cellranger中自带了aggr的整合功能,而这篇文章(Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA-sequencing)的作者也正是这么做得到的组合后的表达矩阵,然后用Read10X读入

关于文章
这是发表在2017年10月的NC文章。
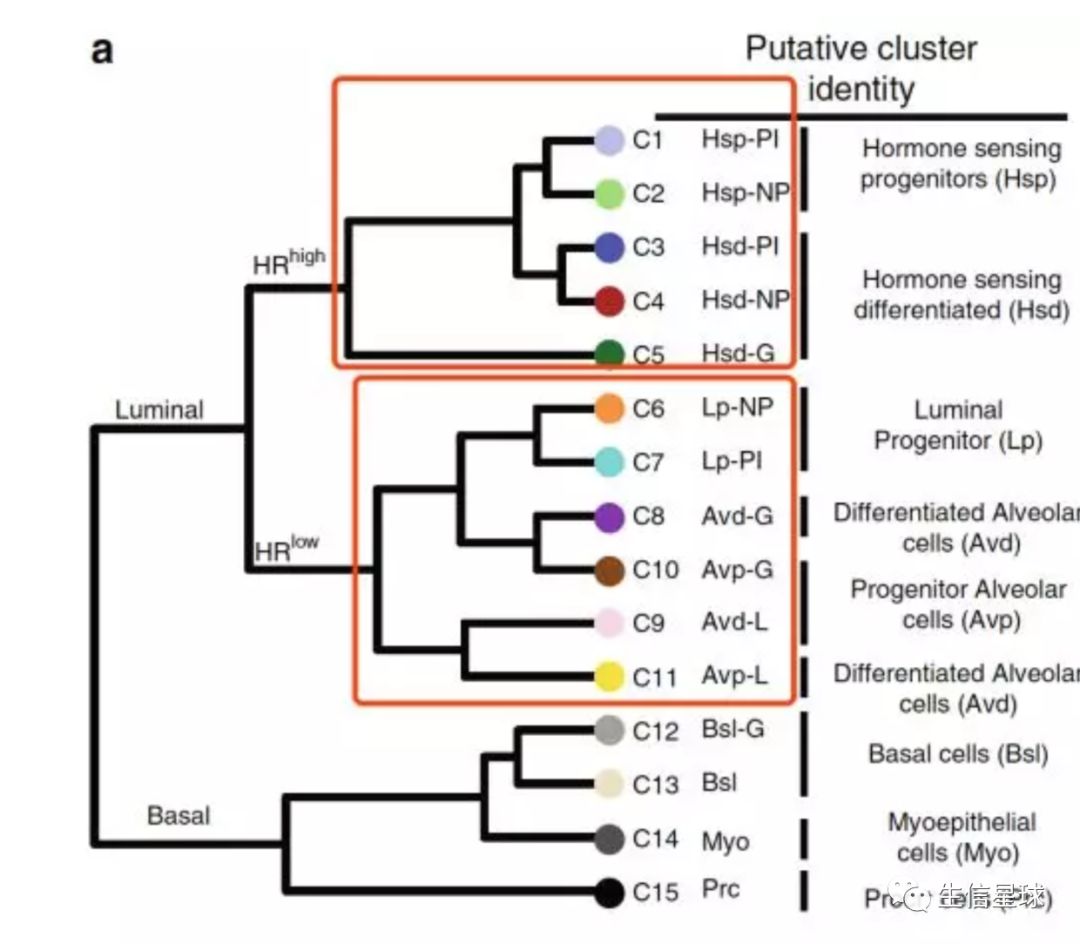
作者的论点是:乳腺上皮细胞对研究乳腺癌的发展很重要,但目前只有很少的marker可以追踪这群细胞,因此有必要探索乳腺发育不同阶段的乳腺上皮细胞变化。
实验涉及了四个时期:8 weeks virgin =》nulliparous (NP) 未怀孕时期、14.5d gestation (G) 妊娠期第14.5天、6d lactation (L) 哺乳期第6天、11d post involution (PI) 完全退化第11天。其中每个时期都采集两只老鼠的组织细胞,所以一共8个样本,然后使用10X建库,那么最后的测序文件就是8*3 = 24个
如果要对这24个文件分别去整合,使用seurat的merge函数即可,不过问题的关键是:如何用代码将这些样本区分开,然后分别构建对象,最后merge这些对象
开始操作
第一步:准备原始测序数据
我们下载第一个:GSE106273_RAW.tar(183.5 Mb) https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE106273&format=file
感觉手机下载速度就是比电脑快,这个文件在手机上下载3分钟,电脑预计时间1小时
下载后解压,整个过程直接在Rstudio中的Terminal直接完成
第二步:整理数据
思路:根据中间的分组信息(NP、G)将包含的文件放到相应的文件夹中
方法一:shell脚本
# 将同一组数据放在同一目录下
ls GSM* | awk -F '_' '{print $2"_"$3}'| uniq | while read i;do mkdir $i;mv *$i*gz $i;done
# 各自重命名
find -name "*barcodes.tsv.gz" | while read i;do mv $i $(dirname $i)/barcodes.tsv.gz;done
find -name "*genes.tsv.gz" | while read i;do mv $i $(dirname $i)/genes.tsv.gz;done
find -name "*matrix.mtx.gz" | while read i;do mv $i $(dirname $i)/matrix.mtx.gz;done
方法二:R脚本
# 列出当前目录下所有开头是GSM的文件
fs=list.files('./','^GSM')
# 然后获取四个样本信息
library(stringr)
samples=str_split(fs,'_',simplify = T)[,1]
# 设置一个循环,对每个样本信息做同样的事:
#(1)找到包含这个样本的文件(用grepl)
# (2)设置对应的目录名(str_split+paste)然后创建目录(用dir.create)
# (3)将文件放到对应目录(采用的是file.rename)并重命名文件
lapply(unique(samples),function(x){
y=fs[grepl(x,fs)]
folder=paste(str_split(y[1],'_',simplify = T)[,2:3],
collapse = '')
dir.create(folder,recursive = T)
file.rename(y[1],file.path(folder,"barcodes.tsv.gz"))
file.rename(y[2],file.path(folder,"genes.tsv.gz"))
file.rename(y[3],file.path(folder,"matrix.mtx.gz"))
})
需要注意的是,Read10X函数需要读取解压后的文件,于是还要对所有的数据文件进行解压
find ./ -name "*gz" |xargs gunzip
常见错误:
- 说找不到Barcode文件,但明明存在Barcode:
Error in Read10X() : Barcode file missing那很有可能是因为三个10X数据的命名出了问题,一定要命名成"barcodes.tsv” “genes.tsv"“matrix.mtx”
【补充:
cellranger的V2版本得到的结果分别是:barcodes.tsv、genes.tsv、matrix.mtx;
V3版本得到的结果分别是:matrix.mtx.gz、features.tsv.gz、barcodes.tsv.gz】
- 说找不到基因文件,那么就要看看测序数据是不是解压后的
Error in Read10X() : Gene name or features file missing
第三步:批量读取成10X对象
Read10X() + CreateSeuratObject()
# 因为Read10X函数需要对目录进行操作,所以先把目录名提取出来
folders=list.files('./',pattern='[12]$')
> folders
[1] "G_1" "G_2" "L_1" "L_2"
[5] "NP_1" "NP_2" "PI_1" "PI_2"
# 然后使用lapply进行循环(看下lapply的帮助文档就知道,它是对列表或向量进行循环,而apply是对数据框或矩阵操作)
library(Seurat)
sceList = lapply(folders,function(folder){
CreateSeuratObject(counts = Read10X(folder),
project = folder )
})
# 此时的sceList仅仅是一个堆砌了8个10X对象的集合,下一步就要真正合并起来
> sceList
[[1]]
An object of class Seurat
27998 features across 2915 samples within 1 assay
Active assay: RNA (27998 features)
[[2]]
An object of class Seurat
27998 features across 3106 samples within 1 assay
Active assay: RNA (27998 features)
第四步:组合
sce.big <- merge(sceList[[1]],
y = c(sceList[[2]],sceList[[3]],sceList[[4]],
sceList[[5]],sceList[[6]],
sceList[[7]],sceList[[8]]),
add.cell.ids = folders,
project = "mouse8")
> table(sce.big$orig.ident)
G_1 G_2 L_1 L_2 NP_1 NP_2 PI_1 PI_2
2915 3106 5906 3697 2249 2127 1500 4306
save(sce.big,file = 'sce.big.merge.mouse8.Rdata') # 保存的数据是1.4G
补充
官网的merge教程在:https://satijalab.org/seurat/v3.1/merge_vignette.html
描述了三种情况
第一种: merge两个seurat对象(原始数据)
需要注意的是,组合数据时需要注明每个数据的名称,使用add.cell.ids参数指定
pbmc.combined <- merge(pbmc4k, y = pbmc8k, add.cell.ids = c("4K", "8K"), project = "PBMC12K")
pbmc.combined
## An object of class Seurat
## 33694 features across 12721 samples within 1 assay
## Active assay: RNA (33694 features)
# 之后的组合数据就会出现列名的标识
head(colnames(pbmc.combined))
## [1] "4K_AAACCTGAGAAGGCCT" "4K_AAACCTGAGACAGACC" "4K_AAACCTGAGATAGTCA"
## [4] "4K_AAACCTGAGCGCCTCA" "4K_AAACCTGAGGCATGGT" "4K_AAACCTGCAAGGTTCT"
table(pbmc.combined$orig.ident)
##
## PBMC4K PBMC8K
## 4340 8381
第二种:merge两个以上(原始数据)
将参数y 设成一个向量,就可以指定其他的数据
pbmc.big <- merge(pbmc3k,
y = c(pbmc4k, pbmc8k),
add.cell.ids = c("3K", "4K", "8K"),
project = "PBMC15K")
unique(sapply(X = strsplit(colnames(pbmc.big), split = "_"), FUN = "[", 1))
## [1] "3K" "4K" "8K"
table(pbmc.big$orig.ident)
##
## pbmc3k PBMC4K PBMC8K
## 2638 4340 8381
第三种:merge归一化、标准化数据
默认情况,只会组合原始数据,但如果有的数据时标准化之后的呢?
其实可以通过一个参数merge.data = TRUE指定
pbmc4k <- NormalizeData(pbmc4k)
pbmc8k <- NormalizeData(pbmc8k)
pbmc.normalized <- merge(pbmc4k,
y = pbmc8k,
add.cell.ids = c("4K", "8K"),
project = "PBMC12K",
merge.data = TRUE)
看看第一种组合raw data和第三种组合normalized data对比:
#################
# raw data
#################
GetAssayData(pbmc.combined)[1:10, 1:15]
## 10 x 15 sparse Matrix of class "dgCMatrix"
##
## RP11-34P13.3 . . . . . . . . . . . . . . .
## FAM138A . . . . . . . . . . . . . . .
## OR4F5 . . . . . . . . . . . . . . .
## RP11-34P13.7 . . . . . . . . . . . . . . .
## RP11-34P13.8 . . . . . . . . . . . . . . .
## RP11-34P13.14 . . . . . . . . . . . . . . .
## RP11-34P13.9 . . . . . . . . . . . . . . .
## FO538757.3 . . . . . . . . . . . . . . .
## FO538757.2 . . . . . . . . . 1 . . . . .
## AP006222.2 . . . . . . . . . . . 1 . . .
#################
# normalized data
#################
GetAssayData(pbmc.normalized)[1:10, 1:15]
## 10 x 15 sparse Matrix of class "dgCMatrix"
##
## RP11-34P13.3 . . . . . . . . . . . . . . .
## FAM138A . . . . . . . . . . . . . . .
## OR4F5 . . . . . . . . . . . . . . .
## RP11-34P13.7 . . . . . . . . . . . . . . .
## RP11-34P13.8 . . . . . . . . . . . . . . .
## RP11-34P13.14 . . . . . . . . . . . . . . .
## RP11-34P13.9 . . . . . . . . . . . . . . .
## FO538757.3 . . . . . . . . . . . . . . .
## FO538757.2 . . . . . . . . . 0.7721503 . . . . .
## AP006222.2 . . . . . . . . . . . 1.087928 . . .
上面的组合多个数据就结束了,接下来是检查组合后的分群结果
首先检查原样本分群结果
# 归一化+标准化(移除了不想要的差异来源nCount_RNA)
sce.big <- NormalizeData(sce.big)
sce.big <- ScaleData(sce.big, vars.to.regress = c('nCount_RNA'),
model.use = 'linear', use.umi = FALSE)
# 默认选2000个HVGs
sce.big <- FindVariableFeatures(object = sce.big,
mean.function = ExpMean,
dispersion.function = LogVMR,
mean.cutoff = c(0.0125,4),
dispersion.cutoff = c(0.5,Inf))
# 降维(PCA+tSNE)
sce.big <- RunPCA(object = sce.big, pc.genes = VariableFeatures(sce.big))
sce.big <- RunTSNE(object = sce.big, dims.use = 1:10)
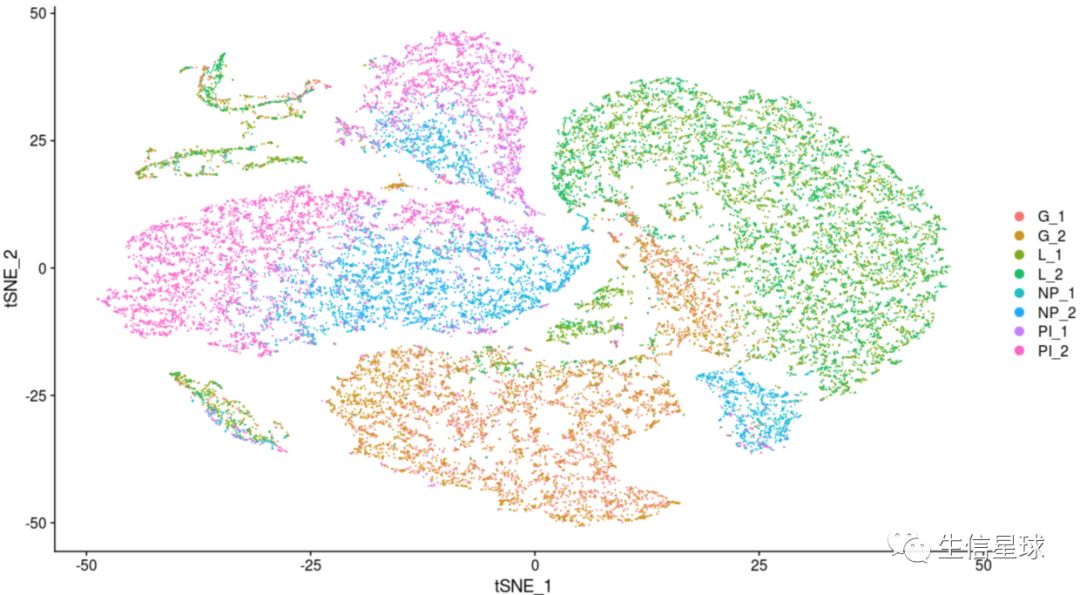
DimPlot(object = sce.big, reduction = "tsne")
# 当然也有ICA的选择
# sce.big <- RunICA(sce.big )
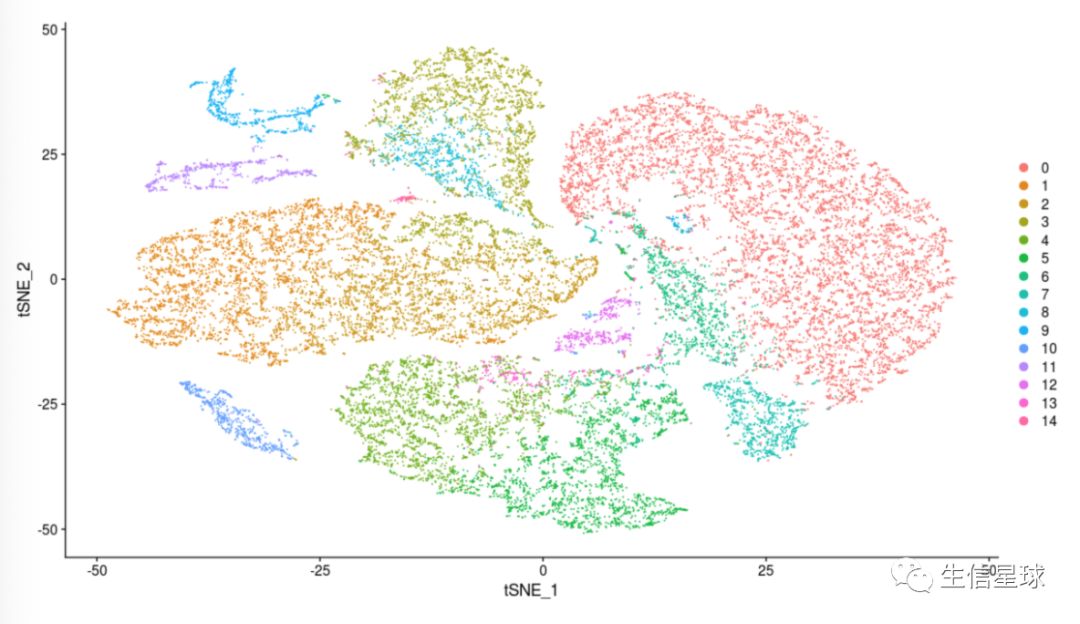
然后鉴定亚群,看看它们的分群结果
ElbowPlot(sce.big)
# 官方建议,下游分析时可以多用几个PCs试试
sce.big <- FindNeighbors(sce.big, dims = 1:20)
# 保持和原文一样的15个亚群
sce.big <- FindClusters(sce.big, resolution = 0.23)
head(Idents(sce.big), 5)
# 新的亚群结果
DimPlot(object = sce.big, reduction = "tsne",
group.by = 'RNA_snn_res.0.23')
# 原样本分群结果
DimPlot(object = sce.big, reduction = "tsne",
group.by = 'orig.ident')
table(sce.big$orig.ident,sce.big@meta.data$RNA_snn_res.0.23)

看到,NP有三个主群、G有三个主群、L有三个主群、PI有两个主群
对比原文的数据
它得到了3个NP、3个G、2个L、3个PI,其余的分给了Basal 4群