scRNA-跟着官网学习单细胞Monocle V3包
刘小泽写于19.8.21+28 这次花费的时间较长,个人感觉这个包比Seurat、Scater难学,说明文档写的实在是太长了!
前言
依旧主打全人工理解(不是翻译)
对这款软件的了解主要是因为它做的发育轨迹、拟时序分析很漂亮,这也是它的主打
Monocle的官网在:
- 版本2:https://cole-trapnell-lab.github.io/monocle-release/docs/#installing-monocle
- 版本3:https://cole-trapnell-lab.github.io/monocle3/monocle3_docs/
首先看新版本的特性和升级之处:
处理的细胞数增加很多(millions of cells)
针对发育生物学领域,做了一些重大改进:
- 发育轨迹的研究流程优化
- 支持
UMAP推断发育轨迹,无缝衔接到
reduceDimension函数,或者使用独立的函数UMAP - 支持多个祖源(toots)的发育推断
- 利用这个算法 approximate graph abstraction 理解各条发育轨迹的分离及平行发育的轨迹
- 3D构建发育轨迹
除了发育轨迹以外,官方还介绍它在亚型发现、差异分析方面表现不错
另外,版本3在持续不断优化,官方称每几个周就会增加一些新功能
其中的算法细节看2019年发表的 Cao & Spielmann et al.
它的主要分析流程是:
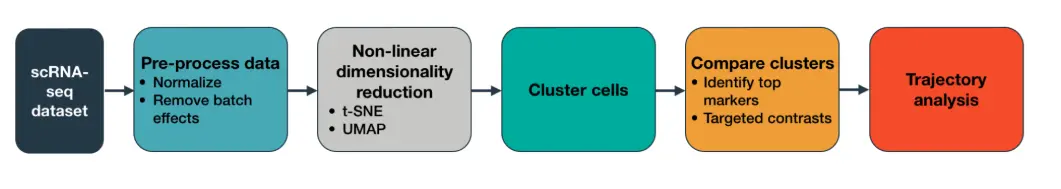
注意:以下是介绍版本3(版本2的使用和3存在一些不同之处,会在”跟着官网学习“这一部分的最后标注)
安装版本3
要求R版本3.5以上,Bioconductor版本3.5以上
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# 一些依赖包
bioc_pkgs <- c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats',
'limma', 'S4Vectors', 'SingleCellExperiment',
'SummarizedExperiment')
for (bpkg in bioc_pkgs){
if (! require(bpkg,character.only=T) ) {
BiocManager::install(bpkg,ask = F,update = F)
require(bpkg,character.only=T)
}
}
# (可选)如果要在细胞聚类时设定分辨率参数(resolution),就要安装一个python包
install.packages("reticulate")
reticulate::py_install("louvain")
# 现在安装3版本,还是要通过github
devtools::install_github('cole-trapnell-lab/monocle3')
# 重启Rstudio,检测是否成功
library(monocle3)
Monocle示例1-细胞聚类及鉴定亚群
创建对象
整体代码就是这样(先了解概况然后下面有实际练习):
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
但是这个对象cell_data_set的设置需要好好理解:
- 这个对象的灵感来自 SingleCellExperiment,因此它的操作也是类似的
expression_matrix是一个行为基因,列为细胞样本的表达矩阵cell_metadata是一个数据框,行为细胞,列是细胞的属性(比如细胞类型、培养环境、培养时间等)gene_metadata是一个数据框,行为feature信息(比如基因),列是基因属性(比如GC含量)【这个很新鲜,因为其他的基于SingleCellExperiment的包如Scater只需要表达矩阵和样本信息即可】。更重要的是,这个数据框中必须有这么一列:gene_short_name,其中保存基因名(简称或Symbol标准名均可,用于作图)- 并且要满足:
expression_matrix的行与gene_metadata的行对应;expression_matrix的列与cell_metadata的行对应
然后测试一下(下载速度会很慢!)
小提示:使用服务器
axel下载,速度会加快(不过这要看服务器的网络配置),如果给力的话,下载均速能到120kb/s,一两分钟就下载好 如果网络真的不给力也没关系,我帮大家下载下来了,在豆豆和花花的公众号”生信星球“后台回复”monocle“就可以得到这三个文件啦:
# 下载数据(如果已经有了,可以跳过)
expression_matrix <- readRDS(url("http://staff.washington.edu/hpliner/data/cao_l2_expression.rds"))
cell_metadata <- readRDS(url("http://staff.washington.edu/hpliner/data/cao_l2_colData.rds"))
gene_annotation <- readRDS(url("http://staff.washington.edu/hpliner/data/cao_l2_rowData.rds"))
# 加载、探索数据
expression_matrix <- readRDS(file = 'cao_l2_expression.rds')
cell_metadata <- readRDS(file = 'cao_l2_colData.rds')
gene_annotation <- readRDS(file = 'cao_l2_rowData.rds')
dim(expression_matrix)
expression_matrix[1:3,1:3]
cell_metadata[1:3,1:3]
head(gene_annotation)
# 创建CDS(Cell Data Set)对象
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
cds
数据预处理
这一步就是告诉Monocle,你想怎么对数据进行归一化、标准化,初步降维是使用PCA(针对标准的RNA-seq,需要指定计算的主成分数)还是LSA(Latent semantic analysis,针对ATAC-seq)。这些都是为后面的聚类操作(tSNE、UMAP做准备)
# 比较耗时
cds <- preprocess_cds(cds, num_dim = 100) #大约2 mins
# 这里设置这么多主成分,可能是因为细胞数太多(4万多个),成分太少不足以代表整体
这个preprocess_cds看下帮助文档就能得知:
- 降维方法
method = c("PCA", "LSI"),默认是PCA - 默认维度
num_dim是50 - 初步降维前归一化
norm_method默认是log处理 - 设置降维方法是PCA时,自动进行标准化处理
检查一下使用的主成分(PCs)是否能够抓取最主要的基因表达变化信息
plot_pc_variance_explained(cds)
# 很像Seurat的ElbowPlot()

可以看到,超过100个主成分后对整体变化的贡献值就没有太多了,而且每多一个PC就会对下游数据分析造成压力
继续降维及可视化
关于维度有必要好好再了解下:
(详见
?reduce_dimension) 那我就编一个”降维“小故事吧 每个细胞中都有2万多基因,可以被当做高维空间上的一个点,而这里说的每个维度就是就不同基因的表达量,也就是说,我们现在有2万个维度上的4万个点;处理的维度越多,后面的轨迹推断也就是越难 好在基因之间不是毫无瓜葛的,各种基因之间总会有错综复杂的联系,通过排除这种”冥冥中的相关性“就会提取出它们之间最不同的地方,更能说明整体的一个特征。于是线性降维就能将2万个维度降到这里的100个,那么之后再处理的话,可能线性降维也有压力。”专人专事:“~于是找来了更专业的非线性降维工具tSNE、UMAP,它们最终能实现再将这100个成分继续压缩,最终得到我们能理解的二维或者3维空间(那么其中包含的样本特征一定是精华中的精华) 英文的解释就是:Each cell can be viewed as a point in a high-dimensional space, where each dimension describes the expression of a different gene.
这里主要采用非线性降维的方法:一种是目前很流行的tSNE,另一种是2018年发布的基于Python的UMAP(UMAP处理大量细胞的算法更优秀;另外与t-SNE相比,UMAP可以保留更多的细胞亚群连续性)
cds <- reduce_dimension(cds, preprocess_method = "PCA",reduction_method = c("UMAP"))
# 大约运行1.5 min
关于这个函数:
多种继续降维的算法:如
"UMAP", "tSNE", "PCA" and "LSI",不过函数默认使用UMAP关于运行加速: 基于
BiocParallel支持多线程运行,使用cores定义;或者使用umap.fast_sgd=TRUE参数可以加速降维 但是自己加速的话,它会友情提示一下:# Note: reduce_dimension will produce slightly different output each time you run it unless you set 'umap.fast_sgd = FALSE' and 'cores = 1'默认得到两个维度
需要定义上一步
preprocess_method使用的算法,默认是LSI
进行可视化:
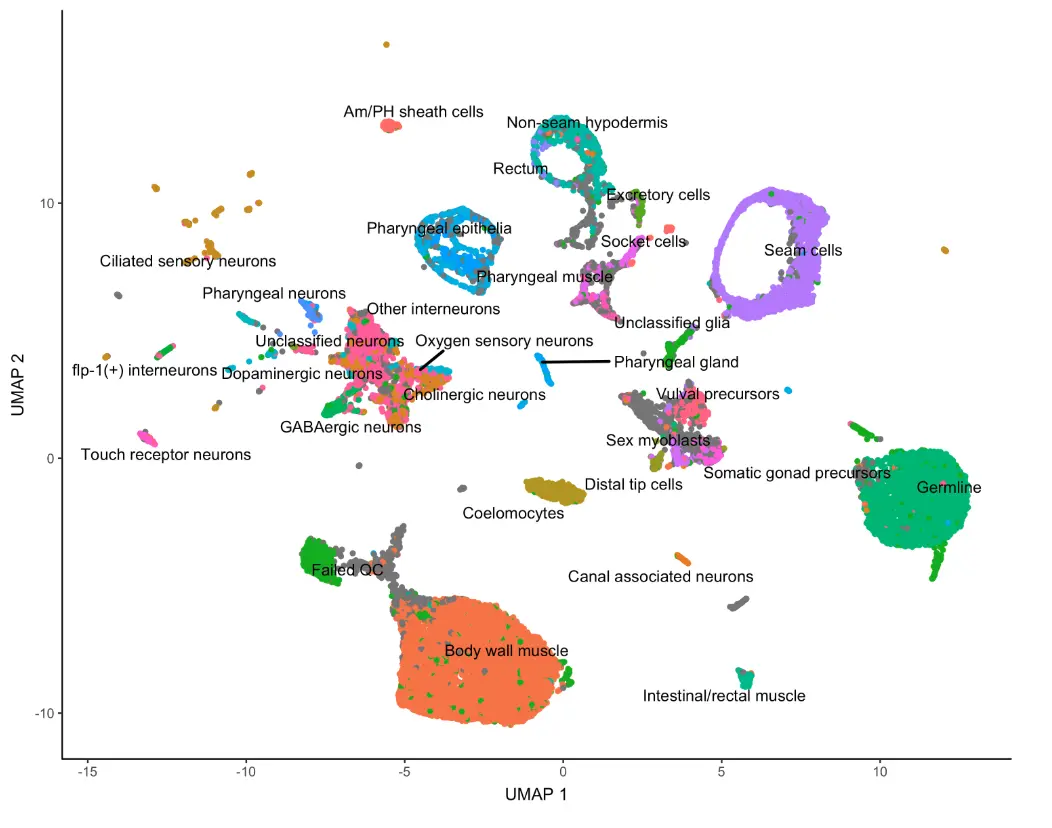
plot_cells(cds)

图中的每个点都代表cds对象中的不同细胞,可以看到不同的细胞分成了不同的群,有的群有几千个细胞,有的群只有少数几个,我们这里直接使用测试数据中做好的细胞标记
# 数据中一共提供了30类细胞(细胞类型及数量见:)
table(colData(cds)$"cao_cell_type")
# 根据这个上色
plot_cells(cds, color_cells_by="cao_cell_type")
# (另外除了用cao_cell_type细胞类型,还能用下面的任意一个)
> colnames(colData(cds))
[1] "plate" "cao_cluster" "cao_cell_type" "cao_tissue" "Size_Factor"
可以看到许多细胞类型再UMAP结果中靠的很近
根据基因表达量进行映射:
plot_cells(cds, genes=c("cpna-2", "egl-21", "ram-2", "inos-1"))

如果想使用tSNE方法降维的话:
cds <- reduce_dimension(cds, reduction_method="tSNE")
# 然后对tSNE结果可视化
plot_cells(cds, reduction_method="tSNE", color_cells_by="cao_cell_type")

其实可以看到,tSNE结果的各个细胞亚群内部的关联性不如UMAP(比如这个stem cells)
检查、移除批次效应 => residual_model_formula_str参数
为了更加真实地反映差异基因的来源是生物学因素,而不是技术因素(比如细胞板批次、细胞培养差别、测序差别等),就首先要检查有没有所谓的”技术批次“对数据差异产生影响。
当进行降维分析时,应该经常检查批次效应,最好在colData(cds) 添加一列,其中注明细胞的批次信息(比如来自哪个细胞板),然后就能根据这个对细胞的批次进行可视化,结果最好是各个细胞亚群的不同批次混在一起,这样我们就能认为:降维所采用的”特异性“的特征并不来自批次
plot_cells(cds, color_cells_by="plate", label_cell_groups=FALSE)

这里看到还不错,但是如果真的担心不同批次会产生其他的影响,可以在降维之前对批次进行校正,保证后面的各个细胞群只来自一个批次,那么它们之间的分群差异就更可能是由于生物因素导致
# 假入要对批次进行校正
cds = preprocess_cds(cds, num_dim = 100, residual_model_formula_str = "~ plate")
# 校正后再降维
cds = reduce_dimension(cds)
# 再次检查批次效应
plot_cells(cds, color_cells_by="plate", label_cell_groups=FALSE)
细胞聚类 => cluster_cells()
这是细胞分群过程中非常重要的一步。Monocle利用
Louvain community detection 这个非监督聚类方法(它是phenoGraph算法的一部分),它和我们常见的Kmeans、hclust层次聚类等还不太一样,这个方法更倾向于找到一个网络中联系紧密的部分,而经常忽略节点的特性;而我们常见的聚类呢,它一般会忽视整体中各个部分的联系,而通过计算两个节点(目标)之间的距离(如欧式距离、曼哈顿距离、余弦相似度等) 找到相似的特性
这一步依赖一个Python模块louvain
关于Python模块在R中的安装: 可以看我写过的 Linux/Mac/Windows的Rstudio安装Python模块总报错,怎么破?
做的话很简单,一个函数,结果保存在了cds@clusters$UMAP$clusters中:
cds = cluster_cells(cds, resolution=c(10^seq(-6,-1)))
plot_cells(cds)
# 这个可视化函数如果不加任何参数,它默认对不同的cluster上色
# 那么有哪些cluster呢?用下面函数查看:
cds@clusters$UMAP$clusters #或者
clusters(cds)
#一共有85个cluster
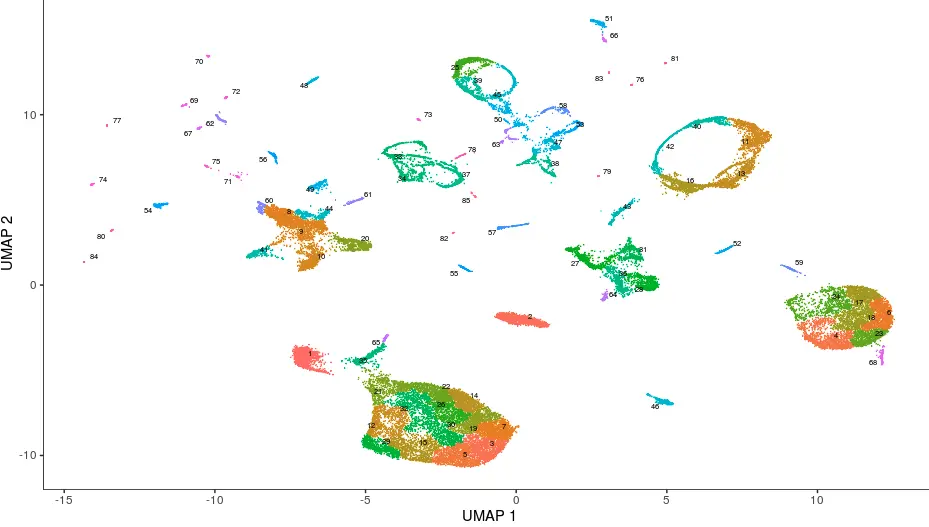
不过这样细分看的太混杂,于是
Alex Wolf et al 利用他们开发的
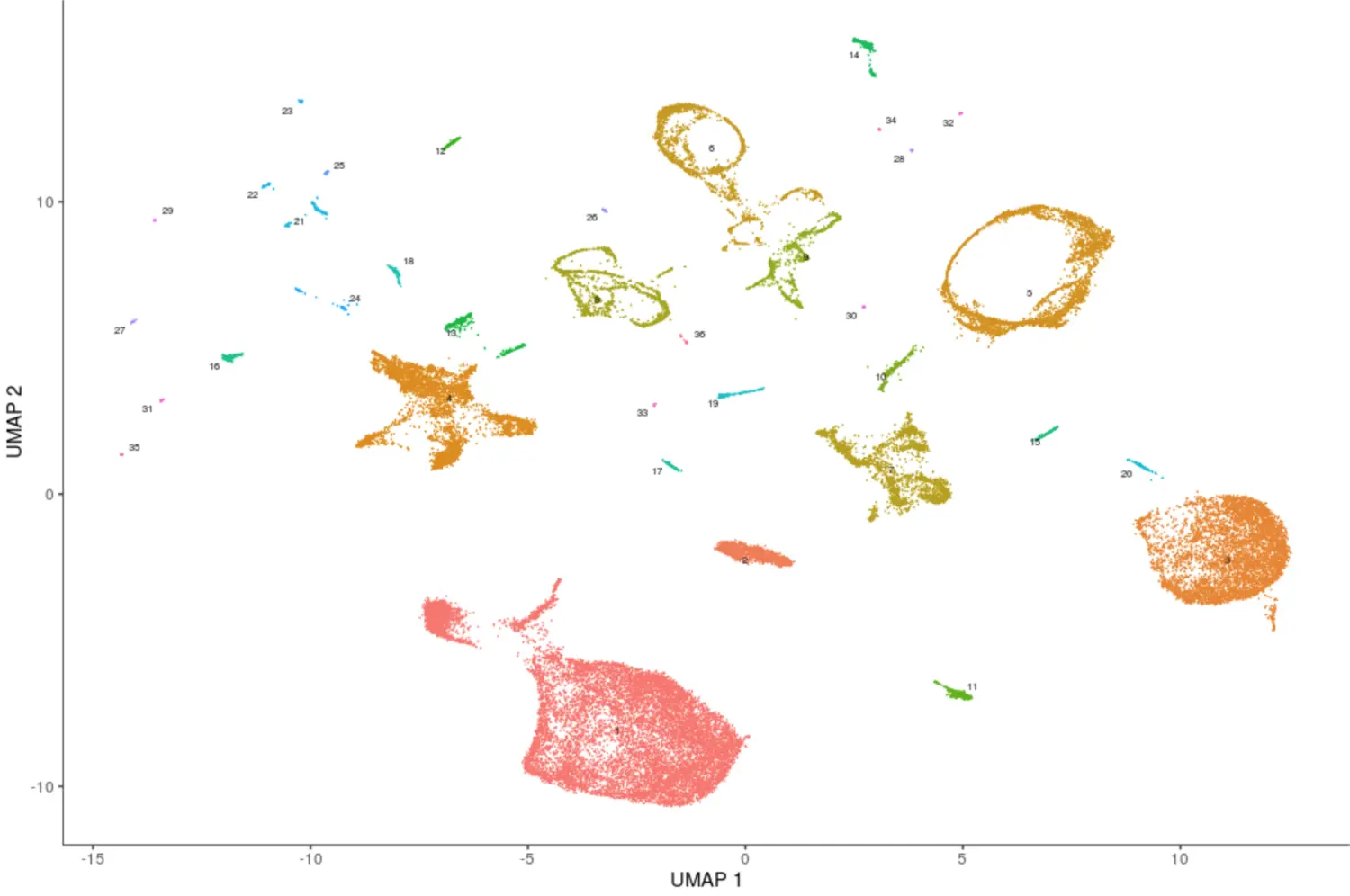
PAGA 算法将一些小的cluster聚合成一个大的partition,结果也是保存在了cluster_cells的计算结果中cds@clusters$UMAP$partitions
plot_cells(cds, color_cells_by="partition", group_cells_by="partition")
# 检查有多少partition
cds@clusters$UMAP$partitions
# 整合成了36个大的partition
除了默认按照cluster编号进行标记不同的亚群,还可以按照每个cluster对应的细胞类型进行可视化:
plot_cells(cds, color_cells_by="cao_cell_type")
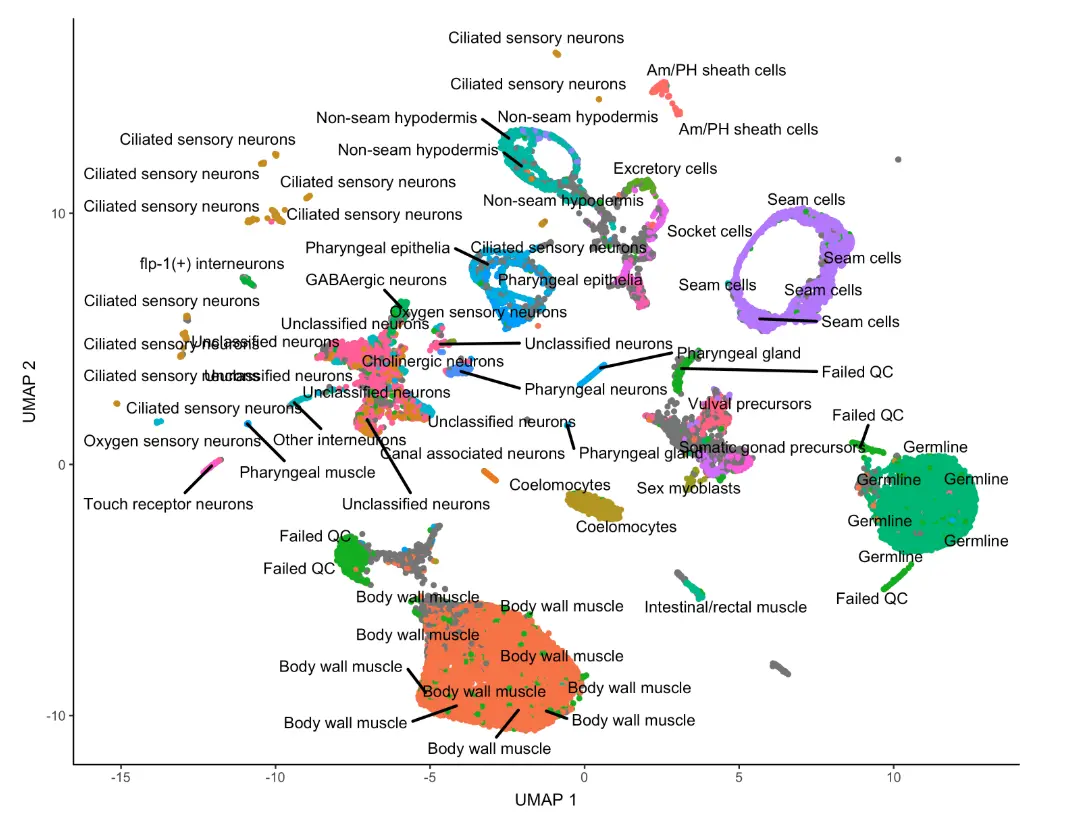
不过这样又很多重复的细胞类型,我们可以对label进行简化:
plot_cells(cds, color_cells_by="cao_cell_type", label_groups_by_cluster=FALSE)
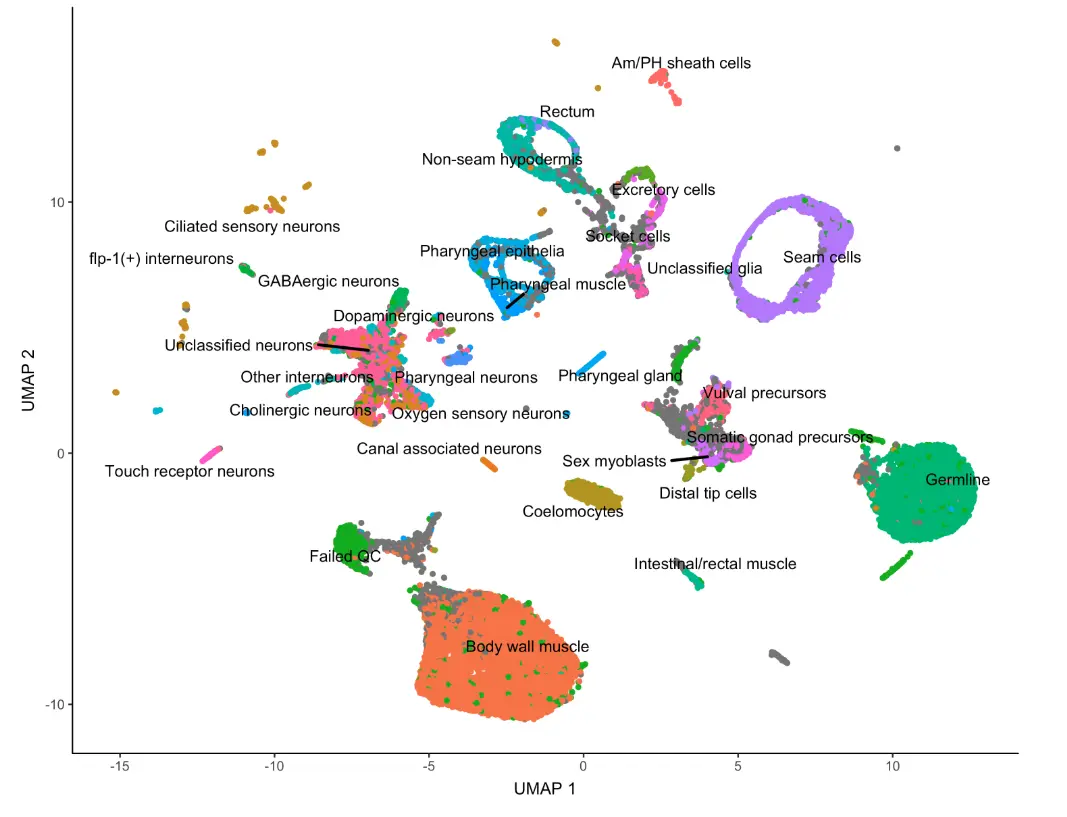
归根结底,都是对细胞的类型、坐标、颜色映射。就是说:
cds@colData$cao_cell_type中存储了每个细胞对应的细胞类型;cds@reducedDims$UMAP中存储了每个细胞UMAP降维后的坐标;cds@clusters$UMAP$partitions或cds@clusters$UMAP$clusters中存储了每个细胞聚类后对应的细胞分群;接下来就是根据坐标对每个细胞进行分群上色、标记类型
找marker基因 => top_markers()
确定了各种亚群以后,我们就想知道什么导致了它们的差异,这就是寻找marker基因的过程,利用top_markers()函数
marker_test_res = top_markers(cds, group_cells_by="partition", reference_cells=1000, cores=8)
# 设置reference_cells是随机挑出来这些数量的细胞作为参照,然后让top_markers和参照集中的基因进行显著性检验;另外reference_cells还可以是来自colnames(cds)的细胞名
marker_test_res[1:4,1:4]
结果得到一个数据框,其中包含了每个partition (因为上面我们设置了group_cells_by)的特异表达基因。
然后我们可以自行过滤:
top_specific_markers = marker_test_res %>%
filter(fraction_expressing >= 0.10) %>%
group_by(cell_group) %>%
top_n(1, pseudo_R2)
# 基因id去重
top_specific_marker_ids = unique(top_specific_markers %>% pull(gene_id))
然后可以对每组的marker基因可视化=> plot_genes_by_group():
plot_genes_by_group(cds,
top_specific_marker_ids,
group_cells_by="partition",
ordering_type="maximal_on_diag",
max.size=3)
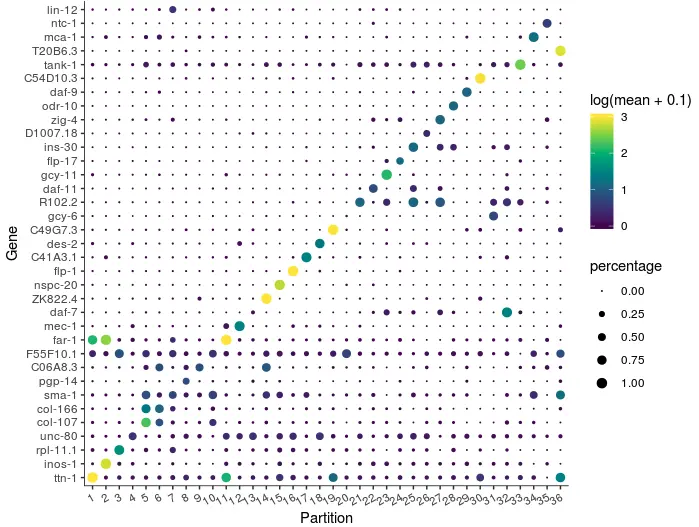
如果要多看几个marker,只需要修改一下top_n
top_specific_markers = marker_test_res %>%
filter(fraction_expressing >= 0.10) %>%
group_by(cell_group) %>%
top_n(3, pseudo_R2)
top_specific_marker_ids = unique(top_specific_markers %>% pull(gene_id))
plot_genes_by_group(cds,
top_specific_marker_ids,
group_cells_by="partition",
ordering_type="cluster_row_col",
max.size=3)

对细胞类型进行注释
因为我们上面的测试数据中给出了细胞类型,所以可以进行聚类后的可视化,但实际上,我们通常只能看到不同的cluster数字和它们的分布(也就是类似于as.character(partitions(cds))的结果),因此利用marker基因去重新定义细胞类型是非常关键的一步。
# 先将partitions的分组由因子型转为字符型
colData(cds)$assigned_cell_type = as.character(partitions(cds))
# 再对字符型重新定义
colData(cds)$assigned_cell_type = dplyr::recode(colData(cds)$assigned_cell_type,
"1"="Body wall muscle",
"2"="Germline",
"3"="Unclassified neurons",
"4"="Seam cells",
"5"="Coelomocytes",
"6"="Pharyngeal epithelia",
"7"="Vulval precursors",
"8"="Non-seam hypodermis",
"9"="Intestinal/rectal muscle",
"10"="Touch receptor neurons",
"11"="Pharyngeal neurons",
"12"="Am/PH sheath cells",
"13"="NA",
"14"="Unclassified neurons",
"15"="flp-1(+) interneurons",
"16"="Canal associated neurons",
"17"="Pharyngeal gland",
"18"="Other interneurons",
"19"="Ciliated sensory neurons",
"20"="Ciliated sensory neurons",
"21"="Ciliated sensory neurons",
"22"="Ciliated sensory neurons",
"23"="Ciliated sensory neurons",
"24"="Ciliated sensory neurons",
"25"="Oxygen sensory neurons",
"26"="Ciliated sensory neurons",
"27"="Unclassified neurons",
"28"="Pharyngeal gland",
"29"="Ciliated sensory neurons",
"30"="Ciliated sensory neurons",
"31"="Ciliated sensory neurons",
"32"="Ciliated sensory neurons",
"33"="Pharyngeal muscle",
"34"="Failed QC")
plot_cells(cds, group_cells_by="partition", color_cells_by="assigned_cell_type")
# Seurat中用RenameIdents 进行细胞类型重定义
# 另外,想从中取子集、过滤的话
cds[,colData(cds)$assigned_cell_type != "Failed QC"]
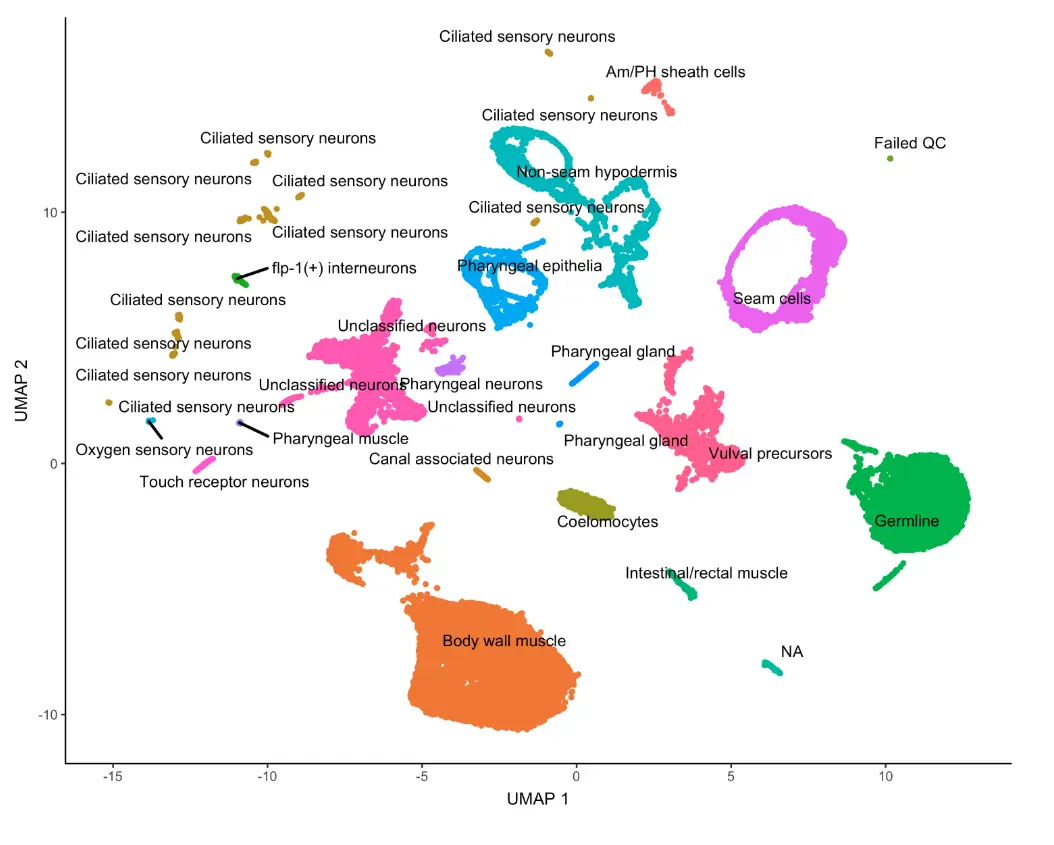
基于Garnett的自动化注释
下面这部分先了解下方法就行,日后有需要再细学
前面对细胞类型进行手动注释有点麻烦,而且cluster编号一旦更改,又要手动去注释一遍。
利用Monocle团队开发的 Garnett ,它可以根据marker基因对细胞进行分类
预处理
# step-1:首先根据上面得到的细胞类型assigned_cell_type,找top_marker
assigned_type_marker_test_res = top_markers(cds,
group_cells_by="assigned_cell_type",
reference_cells=1000,
cores=8)
# step-2:过滤(阈值自定义)
garnett_markers = assigned_type_marker_test_res %>%
filter(marker_test_q_value < 0.01 & specificity >= 0.5) %>%
group_by(cell_group) %>%
top_n(5, marker_score)
# step-3:去重复
garnett_markers = garnett_markers %>% group_by(gene_short_name) %>%
filter(n() == 1)
# step-4:生成marker文件
generate_garnett_marker_file(garnett_markers, file="./marker_file.txt")

得到这个文件,其实也只是自动化实现的第一步,我们自己可以根据已有的知识在其中增加或删除一些marker信息;另外如果发现多个细胞类型中的marker基因有大量重合,就要考虑这几种细胞类型可能都是某一种的细胞的亚型。还有很多关于marker文件修改的帮助信息:Garnett documentation
方法一:自己使用Garnett
# 安装Garnett
devtools::install_github("cole-trapnell-lab/garnett", ref="monocle3")
library(garnett)
# 安装物种注释库(这里的物种是C. elegans秀丽隐杆线虫),目的是为了做基因ID转换
BiocManager::install("org.Ce.eg.db")
colData(cds)$garnett_cluster = clusters(cds)
worm_classifier <- train_cell_classifier(cds = cds,
marker_file = "./marker_file.txt",
db=org.Ce.eg.db::org.Ce.eg.db,
cds_gene_id_type = "ENSEMBL",
num_unknown = 50,
marker_file_gene_id_type = "SYMBOL",
cores=8)
# 这样自己就做了一个分组数据集,官方也建议将高质量的训练集汇总到:https://github.com/cole-trapnell-lab/garnett/issues
#
cds = classify_cells(cds, worm_classifier,
db = org.Ce.eg.db::org.Ce.eg.db,
cluster_extend = TRUE,
cds_gene_id_type = "ENSEMBL")
plot_cells(cds,
group_cells_by="partition",
color_cells_by="cluster_ext_type")
方法二:使用别人提供的分组数据集
例如作者提供了已经做好的数据集(https://cole-trapnell-lab.github.io/garnett/classifiers/ceWhole),我们直接下载就能使用:
load(ceWhole)
cds = classify_cells(cds, ceWhole,
db = org.Ce.eg.db,
cluster_extend = TRUE,
cds_gene_id_type = "ENSEMBL")
Monocle示例2-构建发育轨迹
意义: 在发育生物学中,细胞整个发育阶段中对刺激做出的不同相应导致了细胞功能状态的转化。不同状态的细胞表达不同的基因,从而产生不同的蛋白、代谢物参与不同的生命过程。伴随着细胞状态的转化,转录信息也在不断更换,有的基因起初沉默后来激活。但是这个事情很难去探索,因为要纯化出不同状态并且稳定的细胞基本是不现实的,而scRNA可以不用通过实验方法的纯化细胞,就能研究细胞生命的各个状态。
这也是Monocle最大的亮点—利用算法去学习每个细胞基因表达量的变化,从而对细胞状态进行推断,推断出整体的一个表达轨迹,那么就可以将每个细胞放在特定位置上;另外有可能一个过程有多个结果,那么在轨迹图上就会有多个分支,代表了细胞分化
上面使用的数据集到此结束,下面发育轨迹会用到一套新的数据集(来自 Packer & Zhu et al. 的线虫整个胚胎发育的时间线数据集,这里主要节选了神经元的数据) 需要注意的是,下面做出的图和官网给出的图是不同的
载入数据,构建对象 => new_cell_data_set()
expression_matrix = readRDS(url("http://staff.washington.edu/hpliner/data/packer_embryo_expression.rds"))
cell_metadata = readRDS(url("http://staff.washington.edu/hpliner/data/packer_embryo_colData.rds"))
gene_annotation = readRDS(url("http://staff.washington.edu/hpliner/data/packer_embryo_rowData.rds"))
# 文件大小(同样可以公众号后台回复"monocle-2"获取)
#|-- [497K] packer_embryo_colData.rds
#|-- [10.0M] packer_embryo_expression.rds
#`-- [226K] packer_embryo_rowData.rds
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
预处理 => preprocess_cds()
和示例一的操作差不多,只是这里根据原作者(Packer & Zhu et al )的方法处理了一下批次效应
cds <- preprocess_cds(cds, num_dim = 100, residual_model_formula_str = "~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading")
降维可视化 => reduce_dimension()
强烈建议用UMAP
cds <- reduce_dimension(cds)
plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = "cell.type")

看不同基因在不同分支的表达量:
ciliated_genes = c("che-1",
"hlh-17",
"nhr-6",
"dmd-6",
"ceh-36",
"ham-1")
plot_cells(cds,
genes=ciliated_genes,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)

细胞聚类 => cluster_cells()
Monocle不认为一组数据中的所有细胞都来自同一个”祖先“,很多实验中,它们会有多个发育轨迹。Monocle会通过聚类来判断细胞是否应该归属同一个发育轨迹。之前介绍的cluster_cells()中,细胞可以按cluster细分,还可以按partition归为大类。
# 这里就用partition聚类
cds <- cluster_cells(cds)
plot_cells(cds, color_cells_by = "partition")
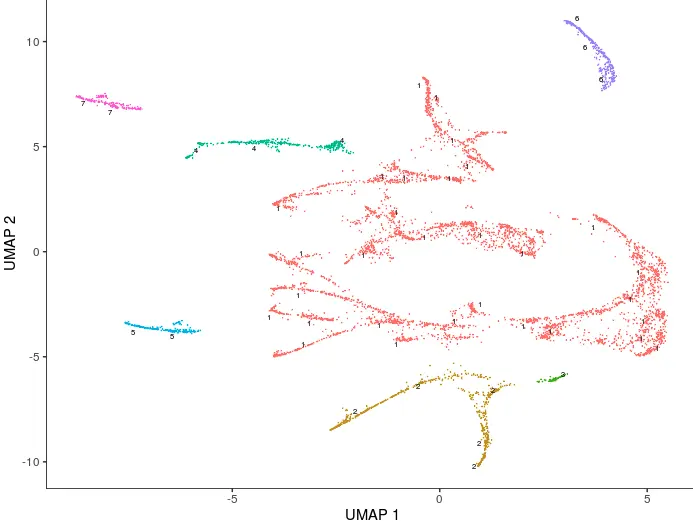
利用learn graph在每个partition中寻找主路径 => learn_graph()
理论上,任何的UMAP或者tSNE降维结果都能找到这样的主路径,选用UMAP是考虑了它能更多展示细胞之间的关联,另外最重要是对数据的了解
cds <- learn_graph(cds)
plot_cells(cds,
color_cells_by = "cell.type",
label_groups_by_cluster=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE)
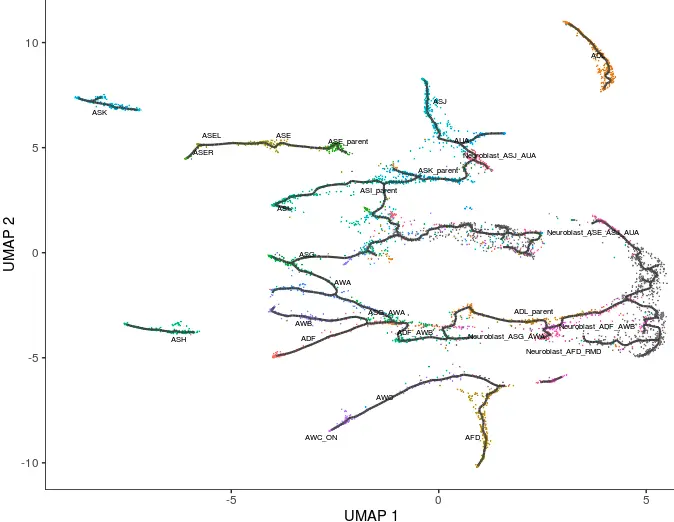
有了初步的轨迹线,接下来就是对细胞出现的先后进行排序 => order_cells()
也就是pseudotime(拟时序分析),推断哪个细胞先出来,哪个细胞后出来。它会对每个细胞在特定的过程(比如分化过程)中的参与度进行评估
很多生命活动中,细胞并非同步发育的,例如在细胞分化阶段,实验捕获到的细胞可能会分布到各个发育过程。因此做单细胞测序时得到的那一群细胞,其中可能包括已经完成某个生命过程的细胞,还可能包括没有开始这个过程的细胞。这种非同步的特性会阻碍我们理解细胞过渡状态究竟发生了怎样的调控。
Monocle的这个算法将基因表达量的变化定义为发育轨迹上生命过程的变化(体现为不同的细胞类型),而不是真正的时间变化(因此我们叫它”拟时序“,而不是”真时间分析“)。
之前得到的轨迹总长度就是细胞从一个阶段的起始到终止所产生的转录水平变化总量;拟时序分析会沿着最短的路径,计算每个细胞和轨迹起始位点的距离
# 先将每个细胞根据”胚胎发育时间区间“进行上色,然后根据胚胎发育过程前后绘制节点
# 在cds@colData中提供了细胞对应的类型以及胚胎发育时间,这个才是真正的关键信息。有了这个信息才能作图。工具不是万能的,它不可能帮助我们去完成生物学中的推断,一定是我们自己先定义好,再交给它进行可视化而已
plot_cells(cds,
color_cells_by = "embryo.time.bin",
label_cell_groups=FALSE,
label_leaves=TRUE,
label_branch_points=TRUE,
graph_label_size=1.5)

- 图中的黑线就是整个架构(注意到整个图并非完全连接的,毕竟是按照partition聚类,每个partition中的细胞差异有点大);
- 浅灰色的圆圈表示”叶片“表示发育轨迹中的不同结局(可以认为拟时序分析的图是一个”根-茎-叶“结构),用参数
label_leaves控制; - 黑色的圆圈表示分支节点,预示着其中的细胞会有不同发展方向,用参数
label_branch_points控制; - 圈中数字大小表示出现时间的先后
从上面的图中我们就能知道出现时间比较早的细胞位置,我们需要对全部的细胞都排个先后顺序,因此要用到order_cells() ,不过这个函数需要一个”根节点“位置,一般是一个partition分配一个根节点。
不过,**怎么简单高效挑选根节点呢?**=> get_earliest_principal_node()
首先根据轨迹图中的节点将与它们最相近的细胞分成组,然后计算来自最早时间点的细胞所占组分,最后挑出包含早期细胞数量最多的节点,认为它是就是根节点
# 官方给出了一个函数,这里定义了一个time_bin,选择了最早的时间点区间。
get_earliest_principal_node <- function(cds, time_bin="130-170"){
# 首先找到出现在最早时间区间的细胞ID
cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)
closest_vertex <-
cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <-
igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names
(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
cds = order_cells(cds, root_pr_nodes=get_earliest_principal_node(cds))
然后进行可视化:
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5)

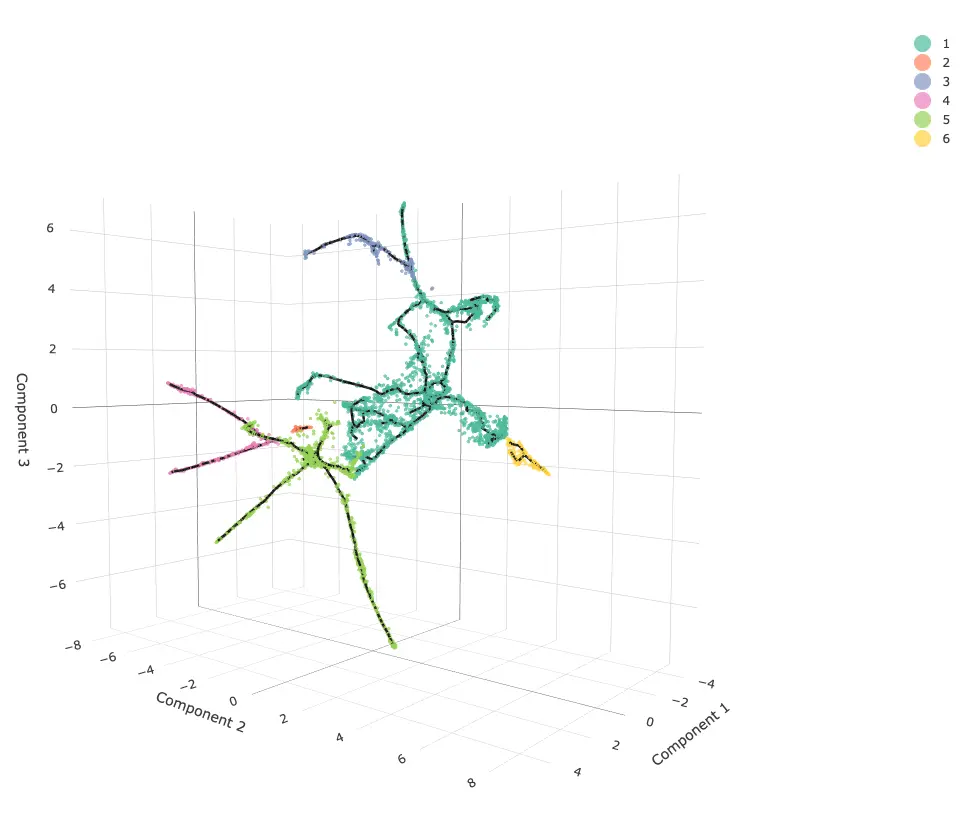
利用3D的发育轨迹对上述内容做个概述
3D轨迹实际上就是降维时选前3个主成分 => max_components = 3,后续都和2D保持类似
需要注意的是,这个3D图需要等待一段时间加载,加载出来的图可以用鼠标拖动
# 3D trajectories(4步走)
cds_3d = reduce_dimension(cds, max_components = 3)
cds_3d = cluster_cells(cds_3d)
cds_3d = learn_graph(cds_3d)
cds_3d = order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))
cds_3d_plot_obj = plot_cells_3d(cds_3d, color_cells_by="partition")
cds_3d_plot_obj
Monocle差异分析
版本3和版本2的差异分析可以说是完全不同,版本3取代了2中的
differentialGeneTest()andBEAM()
Monocle3提供了不同细胞类型之间寻找差异基因的方法,主要有两种:
- Regression analysis:利用
fit_models(),用来评价基因表达是否会受到诸如时间、处理等的影响 - Graph-autocorrelation analysis:利用
graph_test(),用来寻找一条轨迹或不同cluster中基因的差异
方法一:Regression analysis => fit_models()
这一部分将会根据几种不同的标准进行差异分析,并且不同复杂程度的分析用时也有较大差别(几分钟甚至几小时)。教程只会针对一小部分基因进行最简单的分析,但实际上它可以处理上千基因
测试小基因集为:
ciliated_genes = c("che-1",
"hlh-17",
"nhr-6",
"dmd-6",
"ceh-36",
"ham-1")
cds_subset = cds[rowData(cds)$gene_short_name %in% ciliated_genes,]
接下来,Monocle会对每个基因拟合一个回归模型,其中会加入实验中的一些因素(比如时间、处理等)。例如,在胚胎相关的单细胞数据中,细胞会在不同的时间点进行收集,这里就可以检测是否有基因在这段时间内的表达量发生了变化,利用fit_models
gene_fits = fit_models(cds_subset, model_formula_str = "~embryo.time")
# 其中model_formula_str就是要比较的分组对象,如果相获得不同的cluster或者partition的差异基因,就用model_formula_str = "~cluster"或者model_formula_str = "~partition";另外还支持添加多个变量,比如考虑到批次效应 model_formula_str = "~embryo.time + batch"
然后看看哪些基因是与时间因素相关的:
fit_coefs = coefficient_table(gene_fits)
# 挑出时间相关的组分
emb_time_terms = fit_coefs %>% filter(term == "embryo.time")
# coefficient_table()默认使用 Benjamini and Hochberg(BH)方法进行了p值的校正,得到了q值
emb_time_terms %>% filter (q_value < 0.05) %>%
select(gene_short_name, term, q_value, estimate)
# 结果数据集中的6个基因有5个是时间相关的差异基因
除此以外,还能画小提琴图: => plot_genes_violin()
plot_genes_violin(cds_subset, group_cells_by="embryo.time.bin", ncol=2) +
theme(axis.text.x=element_text(angle=45, hjust=1))

如果要考虑批次效应
gene_fits = fit_models(cds_subset, model_formula_str = "~embryo.time + batch")
fit_coefs = coefficient_table(gene_fits)
fit_coefs %>% filter(term != "(Intercept)") %>%
select(gene_short_name, term, q_value, estimate)
模型做出来了,效果到底如何? => evaluate_fits()
对模型进行评价,使用evaluate_fits()
gene_fits = fit_models(cds_subset, model_formula_str = "~embryo.time")
evaluate_fits(gene_fits)
那么模型中到底要不要考虑批次呢?使用compare_models()比较判断。结果会返回一个likelihood ratio test结果,
time_batch_models = fit_models(cds_subset,
model_formula_str = "~embryo.time + batch",
expression_family="negbinomial")
time_models = fit_models(cds_subset,
model_formula_str = "~embryo.time",
expression_family="negbinomial")
compare_models(time_batch_models, time_models) %>% select(gene_short_name, q_value)
其中第一个time_batch_models叫做full model,这个模型知道细胞的收集时间和批次,来预测其中每个基因的表达;第二个time_models叫做reduced model,这个模型只知道细胞的收集时间。因为第一个模型中信息更丰富,所以它对每个细胞中基因表达预测也更准。而Monocle要回答的问题是:第一个模型相比于第二个,究竟做了多少提升? 如果提升的部分大多来自第一个模型的批次效应,那么得到的likelihood ratio test结果就越显著
gene_short_name q_value
<chr> <dbl>
1 ham-1 3.74e-20
2 dmd-6 7.11e-37
3 hlh-17 1.04e- 5
4 nhr-6 5.95e- 3
5 ceh-36 6.23e-10
6 che-1 5.06e-6
结果看到,所有基因的likelihood ratio tests结果都是极显著,说明了数据中的确存在显著的批次效应,也验证了添加批次信息的可靠性。
上面👆所使用的线性模型方法要指定一个基因表达量的分布,大多数研究使用负二项分布,这种分布往往是符合测序reads或UMI count值的,这个方法也被应用在RNA-seq分析(如DESeq2)中
fit_models()函数支持多种分布,默认是:quasipoisson ,它和负二项分布相似,只不过比负二项分布准确性低一点、速度会更快,对于细胞数量多更适用。
关于其他的分布(使用expression_family参数设置):

方法二:Graph-autocorrelation analysis => graph_test()
注意:下面的代码和图片看看就好(因为不能运行)
# 选一部分神经元细胞
neurons_cds = cds[,colData(cds)$assigned_cell_type == "Neurons"]
plot_cells(neurons_cds, color_cells_by="partition")

从图中可以看到,存在不同类型的神经元细胞,它们也许来自不同亚群。为了探索这些不同类型的细胞之间是哪些基因差异表达,一种方法就是上面的线性回归。不过对于UMAP或tSNE的图,graph_test()这个函数可以进行一个叫做
Moran’s I 的spatial autocorrelation分析方法
Moran's I这个指标叫:莫兰指数,澳大利亚统计学家莫兰于1950年提出。它是用来度量空间相关性的一个指标,数据经过方差归一化之后,会落在-1.0和1.0之间。这个值大于0表示空间正相关,值越大空间相关性越强;等于0时空间是随机性分布;小于0时表示空间负相关,值越小空间差异越大
pr_graph_test_res = graph_test(neurons_cds, neighbor_graph="knn", cores=8)
pr_deg_ids = row.names(subset(pr_graph_test_res, q_value < 0.05))
pr_graph_test_res结果是一个数据框,存储了cds对象中每个基因的Moran's I 检验结果。正值表示基因分布在UMAP空间的一小块”焦点“区域(例如一个或几个cluster)。
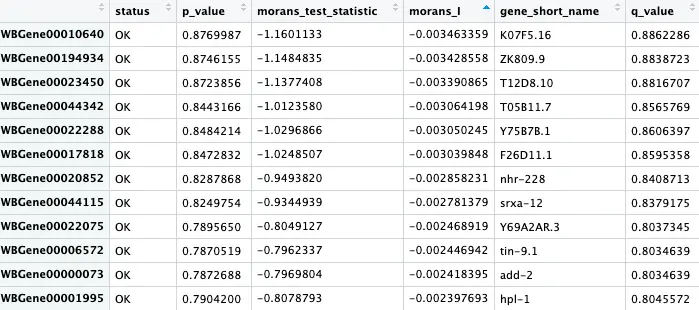
现在有了和”焦点“区域和基因,那么怎么把二者结合起来?
将共同作用的基因合并成模块 => find_gene_modules()
根据这些共同作用基因的表达量对所有细胞进行UMAP,然后利用Louvain community analysis将它们聚集成小模块,然后将小模块和大cluster甚至更大的partition联系起来
gene_module_df = find_gene_modules(neurons_cds[pr_deg_ids,], resolution=1e-2)
得到的结果gene_module_df中有一行记录了每个基因和module的对应关系
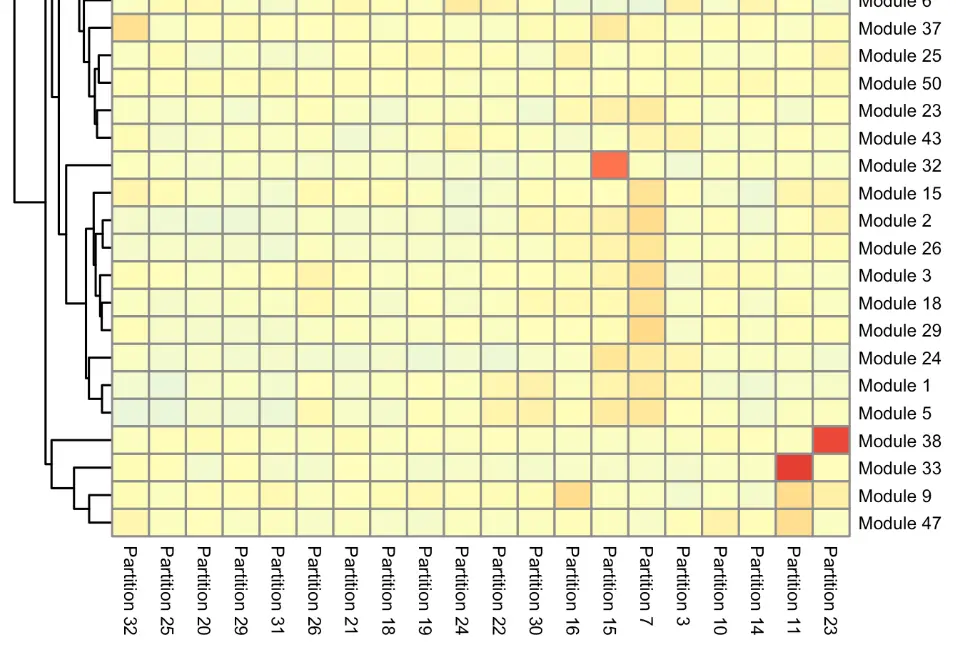
要看哪个module和哪个cluster/partition相关,有两种可视化方法:
第一种:
cell_group_df = tibble::tibble(cell=row.names(colData(neurons_cds)), cell_group=partitions(cds)[colnames(neurons_cds)]) agg_mat = aggregate_gene_expression(neurons_cds, gene_module_df, cell_group_df) row.names(agg_mat) = stringr::str_c("Module ", row.names(agg_mat)) colnames(agg_mat) = stringr::str_c("Partition ", colnames(agg_mat)) pheatmap::pheatmap(agg_mat, cluster_rows=TRUE, cluster_cols=TRUE, scale="column", clustering_method="ward.D2", fontsize=6)结果大体是这样:
第二种:针对少数量的module可以看得比较清楚,这里选了4个
plot_cells(neurons_cds, genes=gene_module_df %>% filter(module %in% c(16,38,33,42)), group_cells_by="partition", color_cells_by="partition", show_trajectory_graph=FALSE)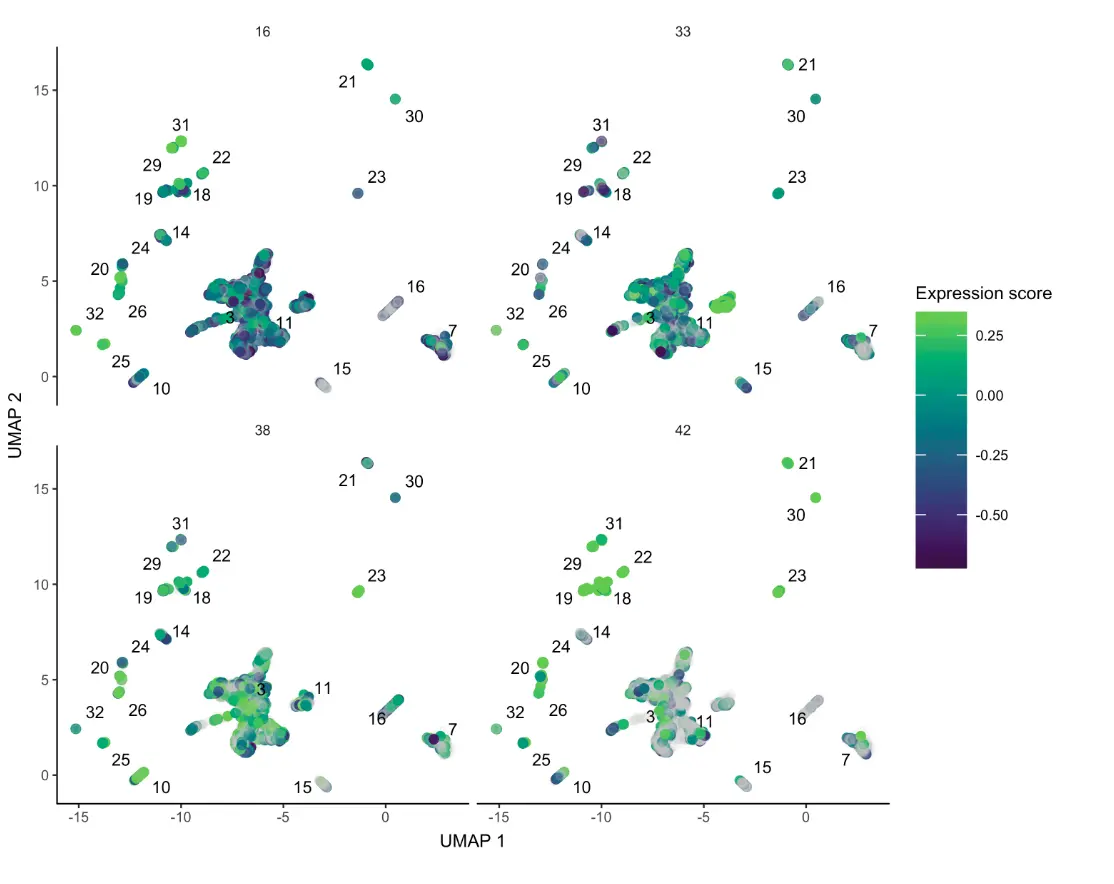
找到影响发育轨迹的基因
这里很重要,但学完后并没有恍然大明白的感觉,感觉跟不上作者的思维了。还要继续摸索这部分
ciliated_cds_pr_test_res = graph_test(cds, neighbor_graph="principal_graph", cores=4)
# 使用neighbor_graph="principal_graph"来检验轨迹相邻的细胞的表达是否相关
pr_deg_ids = row.names(subset(ciliated_cds_pr_test_res, q_value < 0.05))
# 从pr_deg_ids中挑选几个基因,然后可视化
plot_cells(cds, genes=c("hlh-4", "gcy-8", "dac-1", "oig-8"),
show_trajectory_graph=FALSE,
label_cell_groups=FALSE,
label_leaves=FALSE)
# 可以将这些在轨迹上变化的pr_deg_ids继续分为小模块
gene_module_df = monocle3:::find_gene_modules(cds[pr_deg_ids,], resolution=c(0,10^seq(-6,-1)))
cell_group_df = tibble::tibble(cell=row.names(colData(cds)), cell_group=colData(cds)$cell.type)
agg_mat = aggregate_gene_expression(cds, gene_module_df, cell_group_df)
row.names(agg_mat) = stringr::str_c("Module ", row.names(agg_mat))
pheatmap::pheatmap(agg_mat,
scale="column", clustering_method="ward.D2")
plot_cells(cds,
genes=gene_module_df %>% filter(module %in% c(29,20, 11,22)),
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)
另外还有一种方法:
先画完整的轨迹图:
plot_cells(cds, show_trajectory_graph=FALSE)
然后挑某一个细胞亚群对应的一段轨迹:
# 假设这里AFD细胞对应一段22、28、35组成的轨迹线
AFD_genes = c("gcy-8", "dac-1", "oig-8")
AFD_lineage_cds = cds[rowData(cds)$gene_short_name %in% AFD_genes,
clusters(cds) %in% c(22, 28, 35)]
plot_genes_in_pseudotime(AFD_lineage_cds,
color_cells_by="embryo.time.bin",
min_expr=0.5)
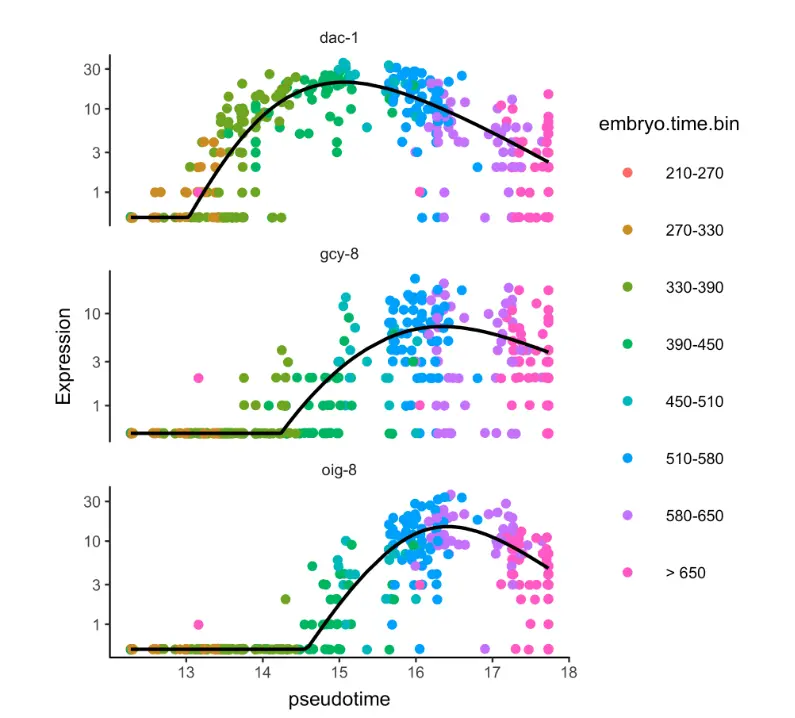
就知道dac-1基因的激活发生在其他两个基因之前
如果使用版本2…
版本2的部分应该是要新写一篇的(后期粘上版本2的链接)
首先创建对象:
- 需要指定
exprs、phenoData、featureData,后两个都要是AnnotatedDataFrame对象(目的是不让用户随便修改,看来还是版本3比较人性化),然后依然是要与表达矩阵行、列对应
#do not run
HSMM_expr_matrix <- read.table("fpkm_matrix.txt")
HSMM_sample_sheet <- read.delim("cell_sample_sheet.txt")
HSMM_gene_annotation <- read.delim("gene_annotations.txt")
pd <- new("AnnotatedDataFrame", data = HSMM_sample_sheet)
fd <- new("AnnotatedDataFrame", data = HSMM_gene_annotation)
HSMM <- newCellDataSet(as.matrix(HSMM_expr_matrix),
phenoData = pd, featureData = fd)
体验
一句话就是:满怀期待,收获满满,看得费劲
优点
- 创建对象变得方便许多,从原来的
new()函数创建对象到现在的数据框,减少了函数的记忆 - 据说是差异分析算法进行了更新,具体新不新有待探索
缺点
- Monocle3的示例数据做的不是很走心,数据下载速度受限。这一点不如Seurat做的清晰明了
- 另外很多地方不能重复出来(比如目前加载的shinny功能还不完善,会出现无响应的状态;文档还有一些小错误)
- 许多代码操作过程复杂
- 版本2、3之间代沟很大,像是完全不同的两个版本,而不是像Seurat2、3几个参数、函数的改变
- 可视化的结果好像比版本2少了一些,不知道这部分是没有改变还是被整合了