scRNA-系统学习单细胞转录组测序scRNA-Seq(四)
刘小泽写于19.3.26 陌生的领域就像一张白纸,怎么画就看自己了
前言
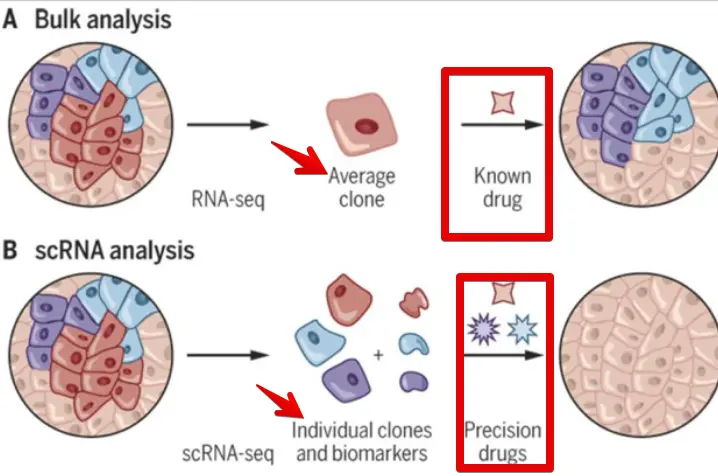
关于单细胞测序,我们已经知道了它和bulk RNA-seq的区别,简单说就是scRNA体现异质性(个体),bulk体现平均程度(总体),因此利用bulk RNA不能区分一个样本中的不同细胞类型

做scRNA的最大的好处就是可以"对症下药”(当然这也是最理想的情况=》什么细胞配合什么种类或者剂量的药物)
** Biomarker:**A biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process, or of a condition or disease. A biomarker may be used to see how well the body responds to a treatment for a disease or condition. Also called molecular marker and signature molecule.
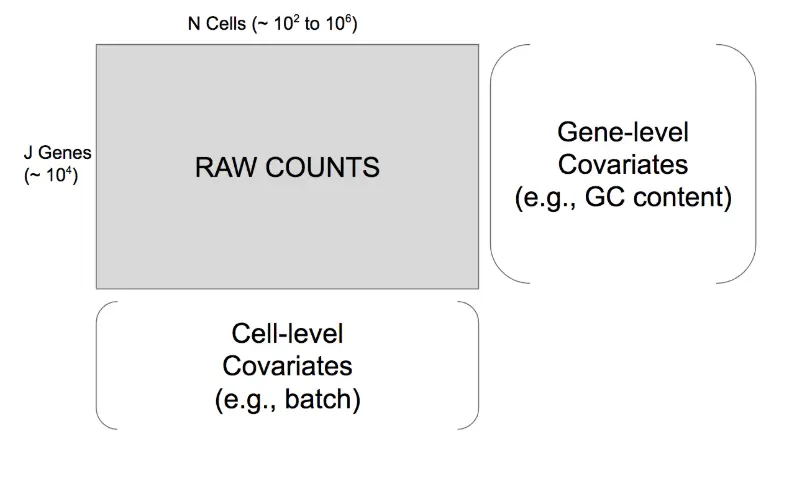
一般在下机后会得到一个表达矩阵,行为基因,列为细胞样本(根据测序平台不同细胞数不同,目前商业化平台有10X和BD,一般都能达到2000-8000细胞量,可以覆盖绝大部分组织的single cell群体类型)。 还包括两个协变量(covariates:解释变量,不为实验者所操纵,但仍影响实验结果):gene-level(比如GC含量)和cell-level(比如细胞测序的批次信息)。
单细胞和bulk转录组表达矩阵主要有两点不同:
bulk的表达矩阵是reads比对到基因的数目;单细胞的表达矩阵是reads比对到某个细胞的某个基因的数目(barcode用来区分细胞;UMI用来区分转录本/基因)
单细胞存在许多的0,有两个可能:第一种是真的基因在细胞中不表达(参与细胞分化的基因并不是在每个cell cycle都表达);第二种是技术误差,基因实际表达但测序没有测到(也就是"dropouts”)
从头开始慢慢理解
这一部分暂时不讨论细节,先看Big Picture
探索表达矩阵
The matrix of counts is the number of sequenced reads aligned to each gene and each sample
假设这个矩阵有10个基因(Lamp5, Fam19a1, Cnr1, Rorb, Sparcl1, Crym, Ddah1, Lmo3, Serpine2, Pde1a) 和5个细胞样本(SRR2140028, SRR2140022, SRR2140055, SRR2140083, SRR2139991)
表达矩阵如下:
> counts
SRR2140028 SRR2140022 SRR2140055 SRR2140083 SRR2139991
Lamp5 10 11 0 0 8
Fam19a1 11 9 0 6 0
Cnr1 0 0 0 12 0
Rorb 0 8 0 0 0
Sparcl1 0 7 0 13 0
Crym 8 9 10 12 5
Ddah1 16 0 0 8 0
Lmo3 0 0 8 13 0
Serpine2 8 0 0 0 0
Pde1a 0 14 8 11 0
# head of count matrix
counts[1:3, 1:3]
# count of specific gene and cell
reads <- counts["Cnr1", "SRR2140055"]
# overall proportion of zero counts
pZero <- mean(counts==0)
# cell library size
libsize <- colSums(counts)
The cell coverage is the average number of expressed genes that are greater than zero in each cell.
目的:ggplot2画出cell coverage
假设目前有一个关于细胞样本的协变量(cell-level),内容如下:
> cell_info
names batch patient
SRR2140028 SRR2140028 1 a
SRR2140022 SRR2140022 1 a
SRR2140055 SRR2140055 2 b
SRR2140083 SRR2140083 2 c
SRR2139991 SRR2139991 2 c
先计算coverage,然后加在cell_info的最后
# find cell coverage 计算count大于0的平均值
# 先将表达量为0的值赋值一个NA,然后计算时忽略掉它们
is.na(counts)=counts==0
coverage <- colMeans(counts,na.rm=T)
cell_info$coverage <- coverage
library(ggplot2)
ggplot(cell_info, aes(x = names, y = coverage)) +
geom_point() +
ggtitle('Cell Coverage') +
xlab('Cell Name') +
ylab('Coverage')
看看大体的流程
测序=》single cell 发展历史
图片来自:Exponential scaling of single-cell RNA-seq in the past decade. (2018 Nature Protocols)
2009-2017不到十年,已报道的细胞研究数目就从1个飞跃至一百万个
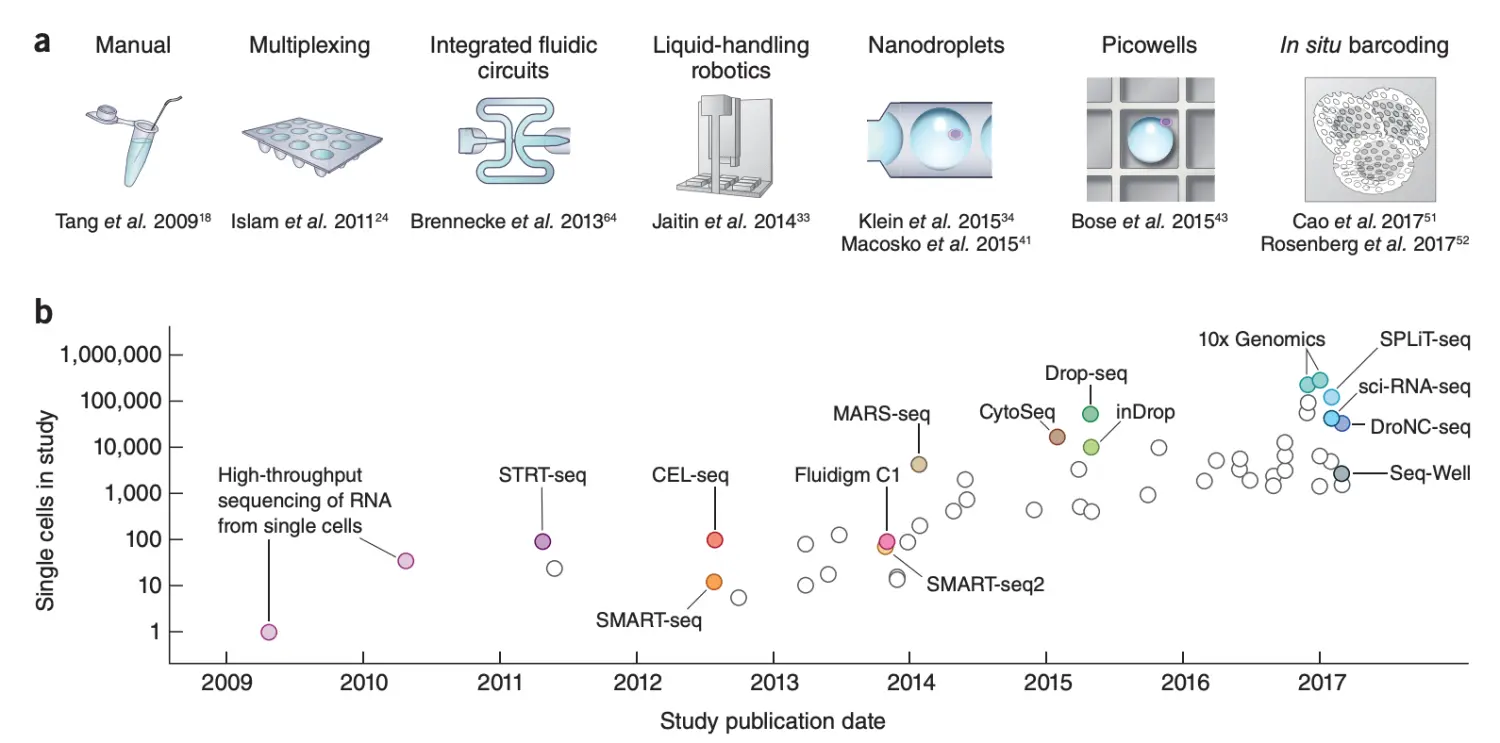
尽管这些方法各不相同,但原理的主要不同之处有两个:分子定量(Quantification)和捕获方法(Capture)
- RNA分子定量(Quantification):主要是两种技术(full-length和tag-based) The full-length:基于全长,试图获取每个转录本覆盖度一致且独特的全长片段; The tag-based:基于标签探针,捕获RNA的5’或3’端序列
- 捕获方法(Capture):不仅决定了细胞的筛选方式以及实验的通量,同时也间接反映了测序外的其他信息。目前主要有三种捕获方法: 微孔 microwell 微流 microfluidic 微滴 droplet
详细看这里:https://hemberg-lab.github.io/scRNA.seq.course/introduction-to-single-cell-rna-seq.html
目前两大商业化单细胞测序公司:
10X Genomics Chromium 利用微流控、油滴包裹和barcode标记实现高通量细胞捕获和标记,一次最多捕获8万细胞。特点就是:细胞通量高**,**建库成本低**,**捕获周期短 主要用于**细胞分型**和**标记因子**的鉴定,从而实现对细胞群体的划分与细胞群体间基因表达差异的检测,此外该技术还可以**预测**细胞分化与**研究**发育轨 迹。
BD Rhapsody 基于微孔平台,通过刚性磁珠进行单细胞捕获(捕获后磁珠可以保存3个月以上);利用成像系统对单细胞分离进行质控。特点是:细胞样本损伤小;质控可视化,监控双细胞比例;细胞捕获较稳定;可以结合蛋白表达量分析转录组;可以设计panel分析特定基因
质控
目的就是减少技术误差,去掉有问题的细胞,一般反映细胞是否有问题主要看两个方面:
- library size:能比对到每个细胞的总reads数(一个ibrary指一个细胞)
- cell coverage:每个细胞中表达基因的平均数
标准Workflow
来自: Bioconductor workflow for single-cell RNA sequencing
可以从下图看到:
第一步就是基因和细胞层次的标准化(normalization):消除与研究无关的生物和技术影响;
第二步是降维,基因数量决定维度,需要从J个基因降到K个基因,实现了两个事情,一是维度降低可以更好掌控数据,二是保留感兴趣的特征同时减少了背景噪音;
第三步是聚类分群,按照降维后的矩阵(K x N,N是细胞数量),这一步给了每个细胞一个cluster number;
第四步是差异分析,找到biomarker,也就是两组细胞的表达差异基因

慢慢翻开细枝末节
加载、创建、获取single-cell数据集
单细胞分析利用的几个包都是高度整合的,里面需要用到许多S4对象,只有理解好这些对象才能看懂分析流程
首先了解一下SingleCellExperiment对象
介绍:
https://www.bioconductor.org/packages/devel/bioc/vignettes/SingleCellExperiment/inst/doc/intro.html
SingleCellExperiment(SCE)是一个S4对象,作者是Davide Risso and Aaron Lun 。官网称它是 light-weight container for single-cell genomics data ,看出它是一个容器,可以装单细胞组学数据。那么就知道和我们日常接触的一般表达矩阵不同,它肯定还包含一些表型信息(对象Object 简单理解就是:包含多个数据框、矩阵或者列表,用到哪个提取哪个。数据框取子集我们用$,对象取子集我们要用@)
之前知道了单细胞数据一般有三个矩阵(表达矩阵raw counts、协变量信息gene-level & cell-level metadata),这个对象就可以将这三个同时存储在一个容器中,另外还有一些其他重要信息,比如:spike-in info,dimentionality reduction coordinates,size factors for each cell
下载、加载包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SingleCellExperiment", version = "3.8")
library(SingleCellExperiment)
创建对象
三个要素:表达矩阵、行名、列名
> # 先设计矩阵(假设counts值服从泊松分布)
> counts <- matrix(rpois(8,lambda = 10),ncol = 2,nrow = 4)
> rownames(counts) <- c("Lamp5","Fam19a1","Cnr1","Rorb")
> colnames(counts) <- c("SRR2140021","SRR2140023")
> counts
SRR2140021 SRR2140023
Lamp5 4 6
Fam19a1 9 13
Cnr1 14 5
Rorb 15 7
# 创建对象第一种方法
#第一个参数assays就是取表达矩阵的list
> sce <- SingleCellExperiment(assays = list(counts = counts),
+ rowData = data.frame(gene = rownames(counts)),
+ colData = data.frame(cell = colnames(counts)))
> sce
class: SingleCellExperiment
dim: 4 2
metadata(0):
assays(1): counts
rownames(4): Lamp5 Fam19a1 Cnr1 Rorb
rowData names(1): gene
colnames(2): SRR2140021 SRR2140023
colData names(1): cell
reducedDimNames(0):
spikeNames(0):
# 创建对象第二种方法(先构建bulk RNA的一个对象SummarizedExperiment,然后转换格式)
> se <- SummarizedExperiment(assays = list(counts = counts))
# as =》 Force an Object to Belong to a Class
> sce <- as(se,"SingleCellExperiment")
上面都是自己创造的模拟数据集,下面看一个真的数据集
来自:Adult mouse cortical cell taxonomy revealed by single cell transcriptomics(2016,Nature Neuroscience)
library(scRNAseq);data(allen)
# 其中包含了379个成年雄性小鼠不同神经元的视觉皮层细胞数据
# allen数据集是一个SummarizedExperiment对象,需要先转换
> sce <- as(allen,"SingleCellExperiment")
> sce
class: SingleCellExperiment
dim: 20908 379
metadata(2): SuppInfo which_qc
assays(4): tophat_counts
cufflinks_fpkm rsem_counts
rsem_tpm
rownames(20908): 0610007P14Rik
0610009B22Rik ... Zzef1 Zzz3
rowData names(0):
colnames(379): SRR2140028
SRR2140022 ... SRR2139341
SRR2139336
colData names(22): NREADS
NALIGNED ... Animal.ID
passes_qc_checks_s
reducedDimNames(0):
spikeNames(0):
# size factors
> sizeFactors(sce) <- colSums(assay(sce))
质控
目的:get out of technical issues and biases
利用的数据集来自:Batch effects and the effective design of single-cell gene expression studies. (2017, Tung et al. )
6 RNA-sequencing datasets per individual : 3 bulk & 3 single-cell (on C1 plates) and each dataset was sequenced in a different batch【Fluidigm C1 platform的低通量的微流控技术,能在捕获细胞时可视化的控制empty wells or doublets】
Three C1 96 well-integrated fluidic circuit (IFC) replicates were collected from each of the three Yoruba individuals.

表达矩阵下载:ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE77nnn/GSE77288/suppl/GSE77288_reads-raw-single-per-sample.txt.gz
library("data.table")
library("dplyr")
reads_raw <- fread("GSE77288_reads-raw-single-per-sample.txt")
setDF(reads_raw)
dim(reads_raw)
# 得到metadata
anno <- reads_raw %>%
select(individual:well) %>%
mutate(batch = paste(individual, replicate, sep = "."),
sample_id = paste(batch, well, sep = "."))
head(anno)
dim(anno)
# 得到表达矩阵
reads <- reads_raw %>%
select(starts_with("ENSG"), starts_with("ERCC")) %>%
t
colnames(reads) <- anno$sample_id
reads[1:5, 1:5]
dim(reads)
# 构建对象
sce <- SingleCellExperiment(assays = list(counts = reads),
rowData = data.frame(gene = rownames(reads)),
colData = anno)
# 去掉样本中表达量均为0的基因
sce2 <- sce[apply(counts(sce), 1, sum) >0,]
dim(sce);dim(sce2)#大约1700个基因不表达
# 添加Spike信息
isSpike(sce2, "ERCC") <- grepl("^ERCC-", rownames(sce2))
> sce2
class: SingleCellExperiment
dim: 18726 864
metadata(0):
assays(1): counts
rownames(18726): ENSG00000237683 ENSG00000187634 ... ERCC-00170
ERCC-00171
rowData names(9): gene is_feature_control ... total_counts
log10_total_counts
colnames(864): NA19098.r1.A01 NA19098.r1.A02 ... NA19239.r3.H11
NA19239.r3.H12
colData names(41): individual replicate ...
pct_counts_in_top_200_features_ERCC
pct_counts_in_top_500_features_ERCC
reducedDimNames(0):
spikeNames(1): ERCC
# Calculate QC metrics
library(scater)
# 常用的feature control:such as ERCC spike-in sets or mitochondrial genes
sce2 <- calculateQCMetrics(sce2, feature_controls = list(ERCC = isSpike(sce2, "ERCC")))
# 结果会在colData这一部分增加许多列,帮助过滤低表达基因和样本
names(colData(sce2))
进行简单的细胞过滤:可以根据**文库大小(Library size)和批次(Batch)**进行
# 1.Filter cells with Library size
# 设置阈值(文库大小基本在2M reads以上,可以设置文库大小的5%作为过滤)
# 阈值的设定没有固定标准,需要考虑具体数据集
threshold <- 100000
# 画密度图
plot(density(sce2$total_counts), main = 'Density - total_counts')
abline(v = threshold)
# 直方图
#####################################
## 一个非常关键的问题:怎么决定哪里作为阈值?
# 理论上细胞分布是遵循泊松分布的“钟形曲线”
# 如果表达量太高(最右侧),可能是一孔两个细胞(doublets);
# 如果表达量太低(最左侧),可能是细胞质量不好或没有细胞
#####################################
hist(
sce2$total_counts,
breaks = 100
)
abline(v=1.3e6, col="red")
# 要保留的细胞
keep <- sce2$total_counts > threshold
# 统计过滤信息
> table(keep)
keep
FALSE TRUE
180 684
# 2.Filter cells with Batch[结果见下图]
# 方法一:
cDataFrame <- as.data.frame(colData(sce2))
# plot cell data
ggplot(cDataFrame, aes(x = total_counts, y = total_counts_ERCC, col = batch)) +
geom_point()
# 方法二:
plotColData(
sce2,
y = "total_counts_ERCC",
x = "total_counts",
colour_by = "batch")
# 要保留的细胞
> filter_by_ERCC <-
sce2$batch != "NA19098.r2" & sce2$pct_counts_ERCC < 25
> table(filter_by_ERCC)
filter_by_ERCC
FALSE TRUE
103 761
> filter_by_MT <- sce2$pct_counts_MT < 30
> table(filter_by_MT)
filter_by_MT
FALSE TRUE
18 846
> sce2 <- filter(sce2,batch != "NA19098.r2")
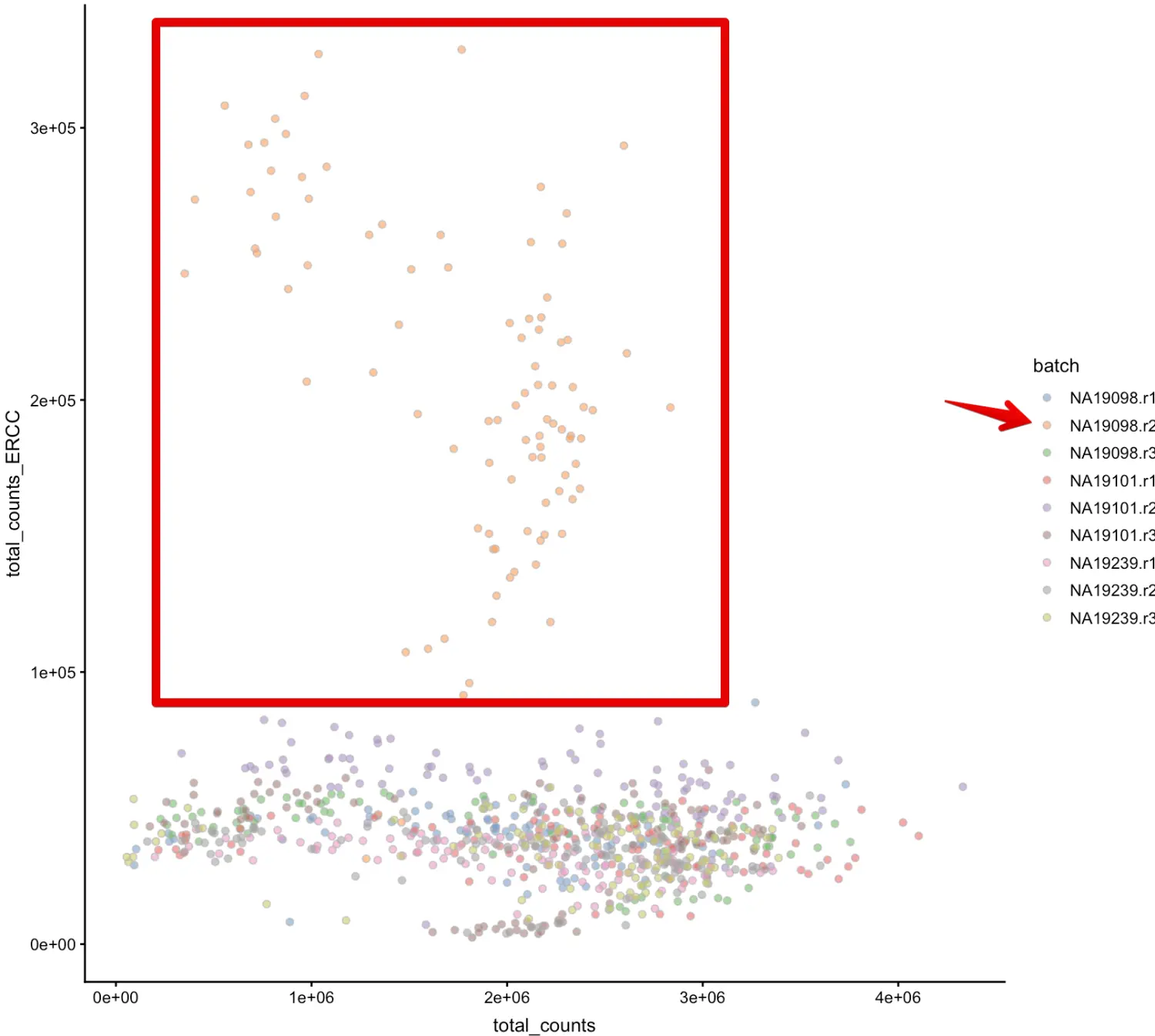
下图中,x轴是total counts,y轴是仅有ERCC基因的total counts,每个点代表一个细胞,细胞根据批次上色。
可以看到**橙色点(r2)**的ERCC占比更高,这个批次在原文中也体现出来了,和其他batch非常不同

ERCC是已知序列和数量的合成mRNA,RNA的读数将提供样本间差异的信息。spike-in control 在确定测序过程中的empty Wells和的dead cells有重要作用,高的ERCC含量与低质量数据相关,并且通常是排除的标准。
细胞过滤后,然后进行基因过滤 (因为有的基因虽然表达,但是却是在被过滤掉的细胞中,因此也算作无效)
目的就是保留至少在1个细胞中表达量不为0
filter_genes <- apply(counts(sce2), 1, function(x){
length(x[x >= 1]) >= 1
})
table(filter_genes)
# 虽然这里不存在低表达基因
sce2 <- sce2[filter_genes,]
下次开始标准化