scRNA-系统学习单细胞转录组测序scRNA-Seq(四+)
刘小泽写于19.3.26 这次主要内容是更新一下第四期的QC部分,另外简单看一下标准化(没有涉及流程),因此标题定了一个 四plus
慢慢翻开细枝末节
上一次说到了加载、创建、获取single-cell数据集、质控(细胞、基因两个层次),继续进行~
上一次质控环节的一些补充说明[主要更新在于过滤更加全面]
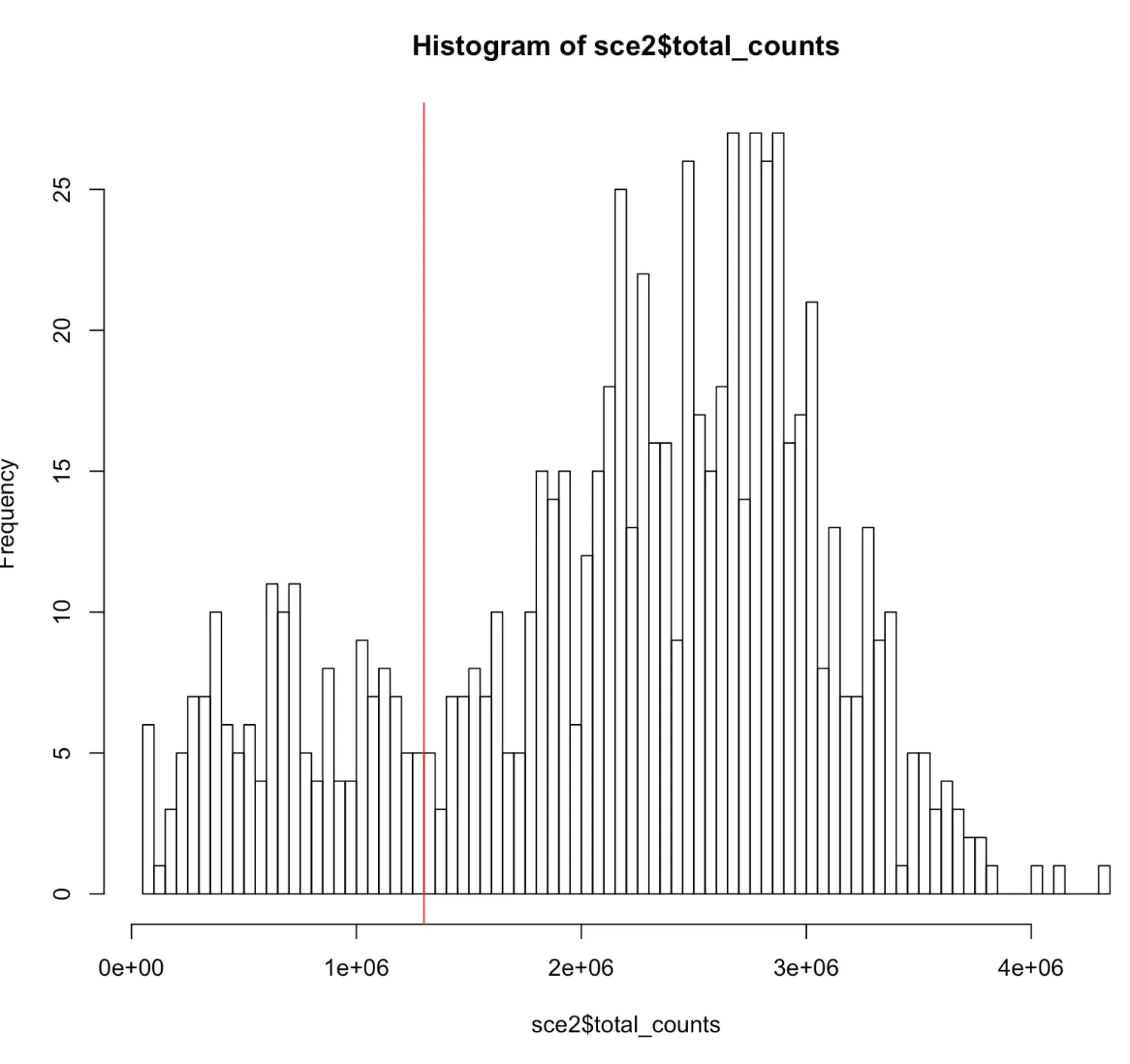
根据文库大小过滤细胞时,我们是根据密度图进行选择,阈值也是按照5%设定的。今天突然想起来,其实还有另一种阈值选择方法,也许更有说服力
# Calculate QC metrics[上一次的代码]
> library(scater)
> sce2 <- calculateQCMetrics(sce2, feature_controls = list(ERCC = isSpike(sce2, "ERCC")))
# names(colData(sce2))
#####################################
# 画个直方图
# 理论上分选细胞是遵循“无限稀释”=》泊松分布的“钟形曲线”
# 如果表达量太高(最右侧),可能是一孔两个细胞(doublets);
# 如果表达量太低(最左侧),可能是细胞质量不好或没有细胞
#####################################
> hist(
sce2$total_counts,
breaks = 100
)
> abline(v=1.3e6, col="red")
# 这样是不是就更直观了呢?
> table(keep)
keep
FALSE TRUE
180 684
除了对总文库大小total_count进行过滤,还可以根据 细胞中检测到的基因数(the number of detected genes in all cells)进行过滤
> hist(
+ sce2$total_features_by_counts,
+ breaks = 100
+ )
> abline(v = 7000, col = "red")
> filter_by_expr_features <- (sce2$total_features_by_counts > 7000)
> table(filter_by_expr_features)
filter_by_expr_features
FALSE TRUE
116 748
# 另外根据ERCC结果可以去掉NA19098.r2这个重复,并且选择ERCC占比低于25的(这个阈值是自定义的)
> filter_by_ERCC <-
sce2$batch != "NA19098.r2" & sce2$pct_counts_ERCC < 25
> table(filter_by_ERCC)
filter_by_ERCC
FALSE TRUE
103 761
一般来讲,QC过滤的方法可以根据:表达的基因数量(total number of unique genes detected in each sample.);文库大小( total number of RNA molecules detected per sample);ERCC、线粒体基因与内源基因的比率(pct_counts_ERCC、pct_counts_MT)[这个比率高说明细胞有可能死亡或者表达受到抑制,即得不到什么表达量]
sce2$use <- (
# sufficient features (genes)
filter_by_expr_features &
# sufficient molecules counted
filter_by_total_counts &
# sufficient endogenous RNA
filter_by_ERCC
# remove cells with unusual number of reads in MT genes
# filter_by_MT
)
上面👆是细胞过滤,下面👇是基因过滤
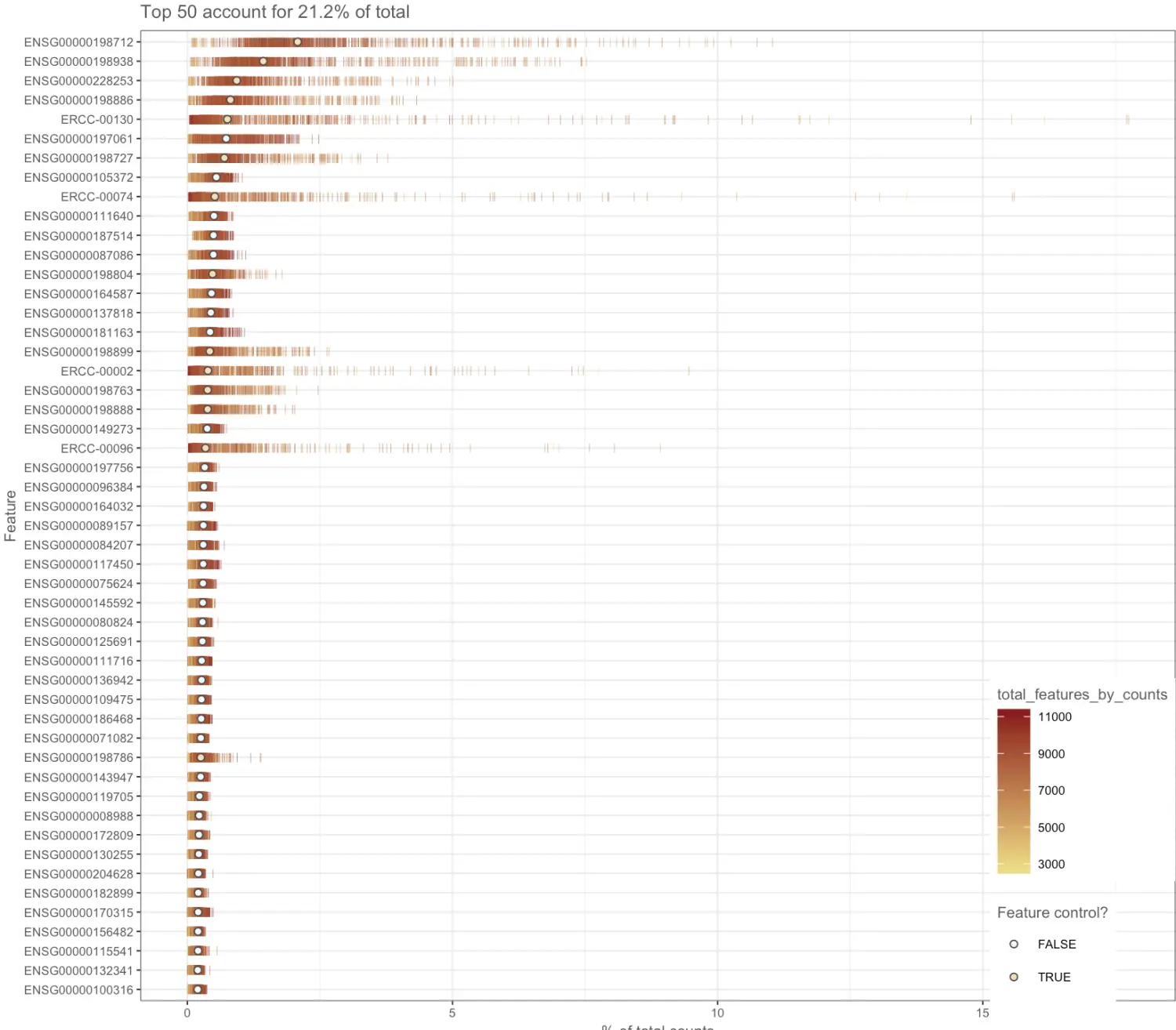
做完QC后,可以进行可视化,检查一下基因表达量(主要看前50),目的是看一下实验设计的spikein如何
The distributions are relatively flat indicating (but not guaranteeing!) good coverage of the full transcriptome of these cells.
plotQC(sce2, type = "highest-expression")
# 结果分布比较平稳表示细胞中全转录组的覆盖度比较好(粗略来看),前15个基因存在一些spike-in(已知浓度的外源RNA分子)[ERCC],因此如果下一步重复实验的话,可以将spikein的比重降低
过滤基因(还是根据至少2个细胞中存在表达量大于1的基因的标准)
filter_genes <- apply(
counts(sce2[ , colData(sce2)$use]),
1,
function(x) length(x[x > 1]) >= 2
)
rowData(sce2)$use <- filter_genes
dim(reads[rowData(reads)$use, colData(reads)$use])
assay(reads, "logcounts_raw") <- log2(counts(reads) + 1)
reducedDim(reads) <- NULL
save(sce2, file = "sce2.RData")
标准化
为何需要标准化?
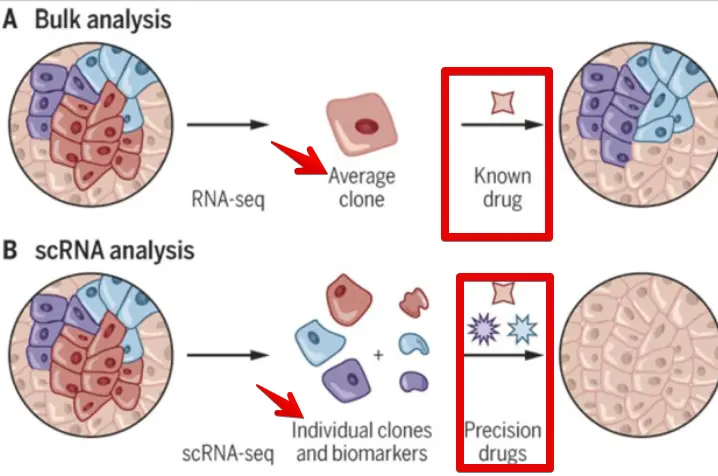
分析单细胞数据其中一个目的就是找到感兴趣的biological signal ,还是以下图为例:
我们可以根据基因表达量将细胞分组,但是这里比较关心的是找到不同细胞亚群的biomarker,然后使用不同的药物治疗。找biomarker的过程并不简单,其中会引入许多的人工技术性误差,以至于掩盖掉真正关心的生物学差异
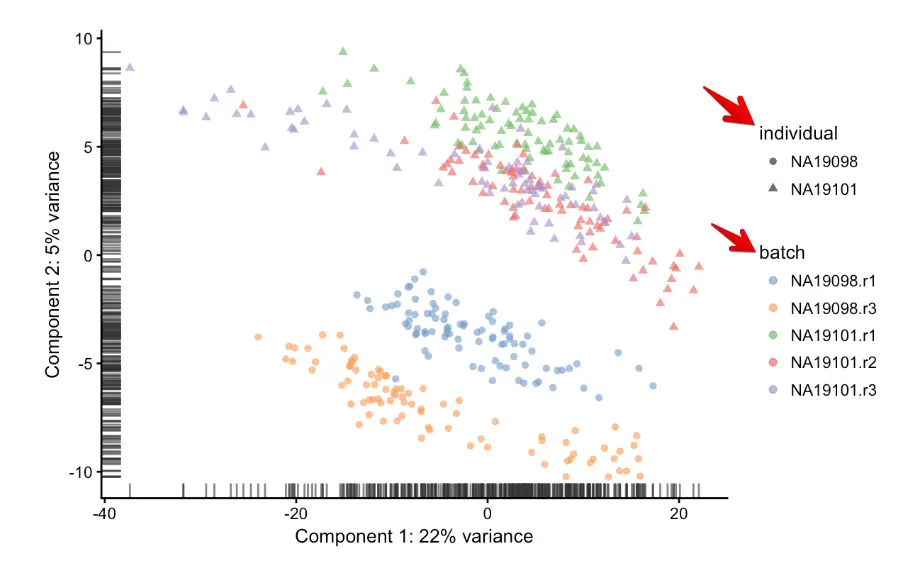
其中一个混杂因素就是”批次效应(batch effect)"。下图中将两组(NA19098和NA19101)进行PCA分析,每个点是一个细胞,细胞被投射到不同维度上,选择其中两个可以代表变化/方差最大的维度来表示数据特征。可以看到,细胞分别按照个体(individual)和批次(batch)进行分组。
按照个体划分是理所当然的,因为我们就是要看看不同个体之间的biological signal,但是按照批次分组就有点说不通了,只是因为细胞是不同批次测序的,而有的批次之间并没有太大的差别,这种情况就属于技术上分组,而不是生物意义上分组。因此,标准化的目的就是:移除不想要的技术差异(例如批次效应),同时保存感兴趣的生物学相关的差异
标准化方法有许多
- 最直接:将表达矩阵的每一列除以标准化因子(normalization factor),这个因子可以是文库大小或者文库大小除以1,000,000(也就是counts per million,CPM);另外像RPKM、TPM都是这种形式的变体
- 其他方法:每个count除以各自不同的归一化因子(scaling factor)。不同方法计算的scaling factor不同,例如
- edgeR中的Weighted trimmed mean of M-values (TMM)
- DESeq中是每个基因在所有细胞中的几何平均数(geometric mean)
- scran中是 zero inflation