099-Bioconductor没想象的那么简单(part4)
刘小泽写于19.4.8
part1:https://www.jianshu.com/p/b030d05a80a4 part2:https://www.jianshu.com/p/c4fdab9929db part3:https://www.jianshu.com/p/f43d9d07a4af
这次看看如何寻找重叠区域(finding overlap)
寻找重叠区域是比较重要的一部分组学分析内容,它将实验结果用比对的reads数据体现出来,然后可以进而推断变异位点,还可以看到基因组上生物学注释区域的不同峰(如基因区域、甲基化区域、染色质状态、进化保守区域等)。对于转录组来说,overlap就是看表达量多少,鉴定不同的isoform
findOverlap热身
目的:看下findOverlap可以干什么,并学习两种模式any和within,了解select参数的使用
两种模式any和within
利用两个IRanges对象和findOverlaps()函数
> library(IRanges)
> qry <- IRanges(start=c(1, 26, 19, 11, 21, 7), end=c(16, 30, 19, 15, 24, 8), names=letters[1:6])
> sbj <- IRanges(start=c(1, 19, 10), end=c(5, 29, 16), names=letters[24:26])
> qry
IRanges object with 6 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
a 1 16 16
b 26 30 5
c 19 19 1
d 11 15 5
e 21 24 4
f 7 8 2
> sbj
IRanges object with 3 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
x 1 5 5
y 19 29 11
z 10 16 7
> hts <- findOverlaps(qry, sbj)
> hts
Hits object with 6 hits and 0 metadata columns:
queryHits subjectHits
<integer> <integer>
[1] 1 1
[2] 1 3
[3] 2 2
[4] 3 2
[5] 4 3
[6] 5 2
-------
queryLength: 6 / subjectLength: 3
找到的overlap结果储存在hts这个对象中,包含两列,分别存储query和subject的索引(可以理解成位置),比如第一行就说明:query的1号(也就是a序列)与subject的1号(也就是x序列有重叠);然后看到queryHits中有2个1出现,那么就是a序列有2段分别和x、z序列有重叠
要获得具体的索引号,分别利用queryHits()和subjectHits()
> names(qry)[queryHits(hts)]
[1] "a" "a" "b" "c" "d" "e"
> names(sbj)[subjectHits(hts)]
[1] "x" "z" "y" "y" "z" "y"
函数找到的重叠区域如下:
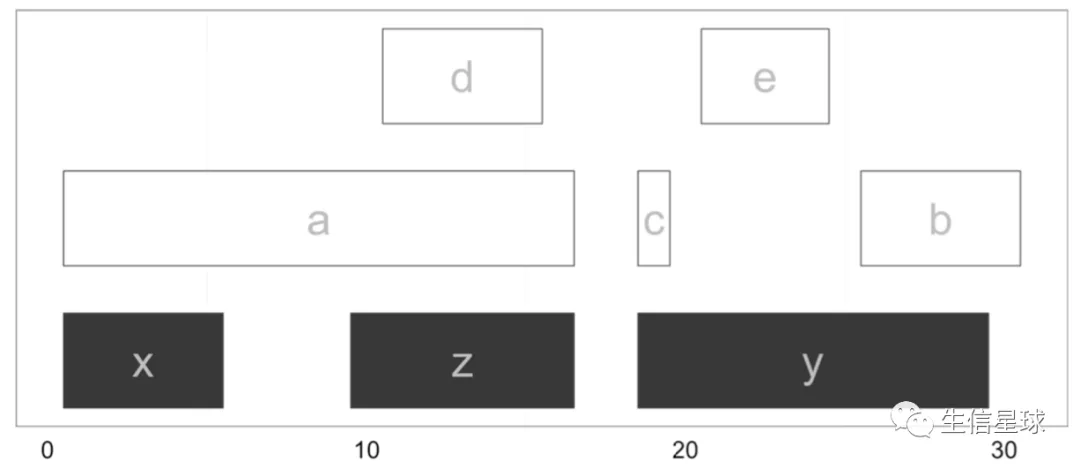
它默认采用any的比对模式,也就是说,只要两个序列有重叠,就计算在内。如果想找query完全落在subject中的序列,就需要使用within模式
> hts_within <- findOverlaps(qry, sbj, type="within")
> hts_within
Hits object with 3 hits and 0 metadata columns:
queryHits subjectHits
<integer> <integer>
[1] 3 2
[2] 4 3
[3] 5 2
-------
queryLength: 6 / subjectLength: 3
这样上图中的a和b就被去掉了,只有c、d、e完全落在z和y中
select参数的使用
解决的问题就是:如果一个qry同时比对到sbj的多个位置,应该选择哪个。影响subjectHits的结果
例如上面的a同时比对到x和z两个位置,默认是使用all 模式,将全部的位置都输出。当然还有其他选择,比如first,last,arbitary 表示输出比对的第一个、最后一个或者随机输出 [如果没有重叠区域,就输出NA]
> hts <- findOverlaps(qry, sbj)
> hts
Hits object with 6 hits and 0 metadata columns:
queryHits subjectHits
<integer> <integer>
[1] 1 1
[2] 1 3
[3] 2 2
[4] 3 2
[5] 4 3
[6] 5 2
-------
queryLength: 6 / subjectLength: 3
> # 使用select参数
> findOverlaps(qry, sbj,select = "first")
[1] 1 2 2 3 2 NA
> findOverlaps(qry, sbj,select = "last")
[1] 3 2 2 3 2 NA
> findOverlaps(qry, sbj,select = "arbitrary")
[1] 1 2 2 3 2 NA
如果有两个gr,分别为grx和gry,如果只想看grx存在的overlap,可以
grx[grx %over% gry]
GenomicRanges
学习它主要因为它比Dataframe效率高多了
它是IRanges(存储基因组区间)的拓展,还包括了染色体信息(seqname)和正负链信息(strand)
https://www.jianshu.com/p/05ac2b781dbf
创建GRanges
> library(GenomicRanges)
Loading required package: GenomeInfoDb
# 起码需要三要素:seqname(可以简写成seq)、ranges、strand
# gr1是最简单的情况
> gr1 <- GRanges(seq = c("chr1","chr2","chr3","chr1"),
+ ranges = IRanges(start = c(5:8),width = 10),
+ strand = "+")
> gr1
GRanges object with 4 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] chr1 5-14 +
[2] chr2 6-15 +
[3] chr3 7-16 +
[4] chr1 8-17 +
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
# 然后gr可以增加其他元素,那些都将作为metadata
gr2 <- GRanges(seq = c("chr1","chr2","chr3","chr1"),
ranges = IRanges(start = c(5:8),width = 10),
strand = "+",
gc = seq(10,70,length.out = 4),
seqlengths = c(chr1=150,chr2=200,chr3=250))
> gr2
GRanges object with 4 ranges and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
[1] chr1 5-14 + | 10
[2] chr2 6-15 + | 30
[3] chr3 7-16 + | 50
[4] chr1 8-17 + | 70
-------
seqinfo: 3 sequences from an unspecified genome
> seqlengths(gr2)
chr1 chr2 chr3
150 200 250
> # 访问seqname
> seqnames(gr2)
factor-Rle of length 4 with 4 runs
Lengths: 1 1 1 1
Values : chr1 chr2 chr3 chr1
Levels(3): chr1 chr2 chr3
> # 访问ranges
> ranges(gr2)
IRanges object with 4 ranges and 0 metadata columns:
start end width
<integer> <integer> <integer>
[1] 5 14 10
[2] 6 15 10
[3] 7 16 10
[4] 8 17 10
> start(gr2)
[1] 5 6 7 8
> end(gr2)
[1] 14 15 16 17
# 这里的width能做什么?=》统计全基因组范围内的基因长度分布
> width(gr2)
[1] 10 10 10 10
# 看一下总共有多少行
> length(gr2)
[1] 4
> # 访问strand
> strand(gr2)
factor-Rle of length 4 with 1 run
Lengths: 4
Values : +
Levels(3): + - *
# 换个行名
> names(gr2) <- letters[1:length(gr2)]
> gr2
GRanges object with 4 ranges and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
a chr1 5-14 + | 10
b chr2 6-15 + | 30
c chr3 7-16 + | 50
d chr1 8-17 + | 70
-------
seqinfo: 3 sequences from an unspecified genome
# 取子集很方便,比如要取overlap的起始位点大于7
> gr2[start(gr2)>7]
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
d chr1 8-17 + | 1
-------
seqinfo: 3 sequences from an unspecified genome
# 看看metadata columns
> mcols(gr2)
DataFrame with 4 rows and 1 column
gc
<numeric>
a 10
b 30
c 50
d 70
# 当然也可以直接选取
> gr2$gc
[1] 10 30 50 70
实用的是,可以直接根据metadata计算一些统计量,这也是GRanges比dataframe处理基因组数据更快的原因之一
比如计算1号染色体的GC content
> mean(gr2[seqnames(gr2) == "chr1"]$gc)
[1] 40
另外还有许多实际应用:
gr2[strand(gr2) == "*"] # 取出不分正负链的
gr2[strand(gr2) != "*"] # 取出分正负链的
gr[gr$score<5 & gr$GC >0.7] #取出gr中score小于5且GC大于0.7
sort(gr2) #按基因组顺序排序(先染色体=》再位置=》正链=》负链=》不分链)
gr2[order(gr2$gc)] #按某一列进行排序(从小到大)
gr2[order(gr2$gc,decreasing = T)] #按某一列进行排序(从大到小)
# 详细可以看:https://www.jianshu.com/p/05ac2b781dbf
flank(gr2,width=2) # 取上游2bp
# 取上游的目的主要是为了找启动子promoter(一般上游2k,下游10)
# 实际上有专门的函数可以做
promoters(gr, upstream = 2000, downstream = 10)
# 使用flank可能会导致上游负数出现,这时把负数变成1就好
start(gr[start(gr)<1])=1
flank(gr2,width=2,start=F) # 取下游2bp(为了分析UTR=》UTR长度不固定=》UTR越长,能够调控的microRNA越长)
# 如果取下游超出了染色体长度,那么在循环中只需要把超过的部分设置成染色体长度
# 一个区域整体平移
shift(gr2,shift=10) # 正负链都向染色体下游平移(正向平移)
shift(gr2,shift=-10) #向染色体上游平移
# reduce只计算一次长度,假设要计算外显子区域长度,肯定存在两个exon交叉的情况,如果直接把每个exon长度相加,那么他们的交叉区域会被记做两次,总长度比实际要大。reduce就可以解决这个问题
# disjoin可以把基因的共同转录区部分去掉,剩下的就是gene specific的内容,研究可变剪切时是很有用的
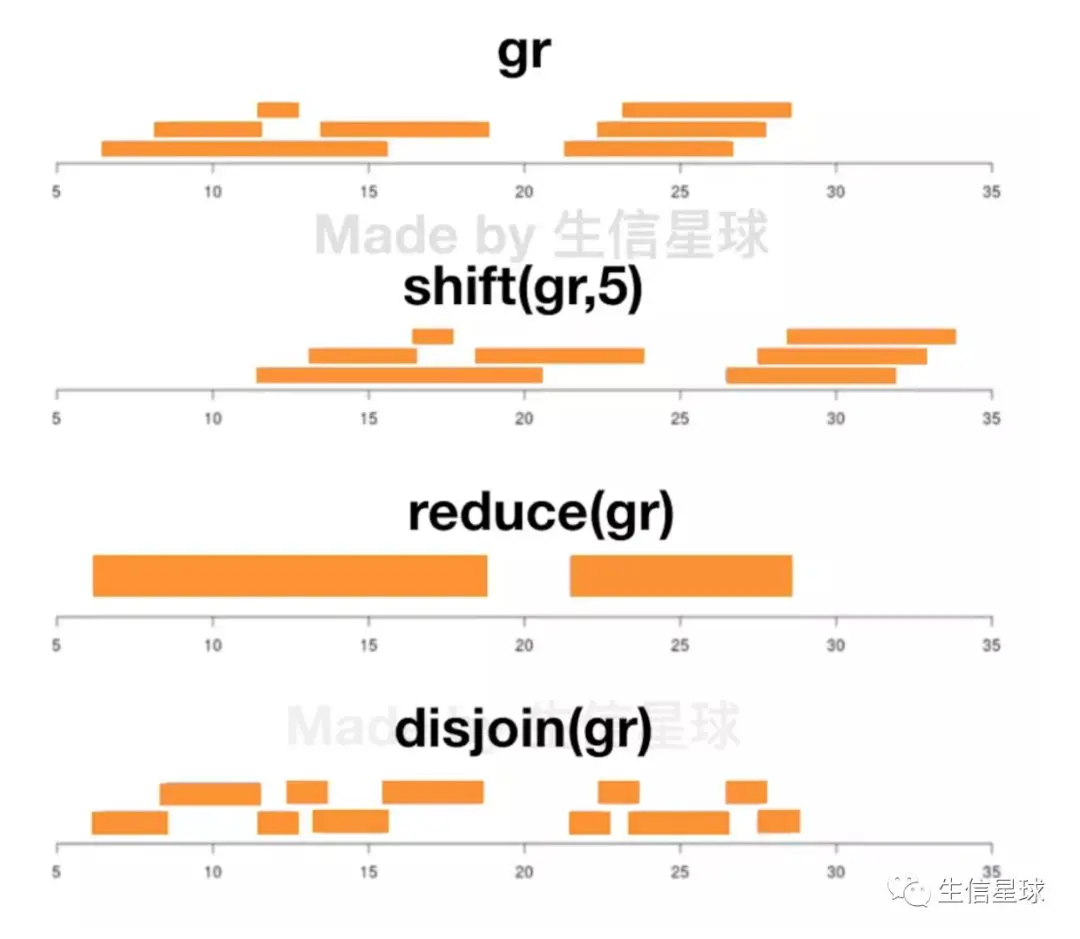
拆分GRanges成GRangesList:
为什么要拆分成list?最重要的原因是,list更适合存储数据,例如现在有一个GRanges对象包含所有的外显子,如果我们想按基因或转录本对这些外显子进行分组,那么在list中就可以很轻松实现
# 这里按照染色体号拆分
> gr_split <- split(gr2,seqnames(gr2))
GRangesList object of length 3:
$chr1
GRanges object with 2 ranges and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
a chr1 5-14 + | 10
d chr1 8-17 + | 70
$chr2
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | gc
b chr2 6-15 + | 30
$chr3
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | gc
c chr3 7-16 + | 50
-------
seqinfo: 3 sequences from an unspecified genome
# 拆分后的子集名称
> names(gr_split)
[1] "chr1" "chr2" "chr3"
# 取第一个子集(chr1
> gr_split[[1]]
GRanges object with 2 ranges and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
a chr1 5-14 + | 10
d chr1 8-17 + | 70
-------
seqinfo: 3 sequences from an unspecified genome
拆分后的补回来,结果还是和gr2一样:
> unsplit(gr_split,seqnames(gr2))
GRanges object with 4 ranges and 1 metadata column:
seqnames ranges strand | gc
<Rle> <IRanges> <Rle> | <numeric>
a chr1 5-14 + | 10
d chr2 6-15 + | 30
b chr3 7-16 + | 50
c chr1 8-17 + | 70
-------
seqinfo: 3 sequences from an unspecified genome
学习GRangesList
这个东西就相当于日常见到的list ,举个例子:
> gr3 <- GRanges(c("chr1", "chr2"), IRanges(start=c(32, 95), width=c(24, 123)))
> gr4 <- GRanges(c("chr8", "chr2"), IRanges(start=c(27, 12), width=c(42, 34)))
> grl <- GRangesList(gr1, gr2)
> grl
GRangesList object of length 2:
[[1]]
GRanges object with 2 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] chr1 32-55 *
[2] chr2 95-217 *
[[2]]
GRanges object with 2 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr8 27-68 *
[2] chr2 12-45 *
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
然后可以unlist ,结果把grl拆分,拆分的gr3和gr4合并在一起
> unlist(grl)
GRanges object with 4 ranges and 0 metadata columns:
seqnames ranges strand
<Rle> <IRanges> <Rle>
[1] chr1 32-55 *
[2] chr2 95-217 *
[3] chr8 27-68 *
[4] chr2 12-45 *
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
可以把多个GRangesList合并,直接c(grl,grl) 即可,本质上它还是个列表(可以使用length查看前后变化),取子集用[],取子集中的元素用[[]]
gr5 <- c(gr2,gr2)
length(gr2) #4
length(gr5) #8
unique(gr5) #结果还是和一个gr2一样
和GRanges一样,GRangesList也可以使用seqnames(),start(),end(), width(), ranges(), strand()等
可以借助循环
# start position of the earliest (leftmost) range
> sapply(gr_split, function(x) min(start(x)))
chr1 chr2 chr3
5 6 7
# number of ranges in every GRangesList object
> sapply(gr_split, length)
chr1 chr2 chr3
2 1 1