089-生信单行命令的美,慢慢体会(一)
刘小泽写于19.3.1
最近一直忙毕业论文,感觉过去的一周像是过去的一个月一样,快接近尾声了,知道我的朋友都知道,我英语单词、配音、Peak连续打卡几百天,现在与其称之为打卡,倒不如对我是一种生活方式,是不做就睡不着觉的事情,我也很开心有这样的一个习惯。因此,虽然这段时间很忙,但是推送也在一直坚持写,anyway,等熬过这段时间就爽了~ 今天在火车上用手机闲来无事,一边听着“潘吉Jenny告诉你”(嗯,是我喜欢的英语播客,也推荐给你哦),一边重新复习了下Shell单行命令
写在前面
文本文件想必是日常中完成一个大的生信计算项目,方法绝对不止一种,但最先想到的肯定是最简单的方法,那么有什么方法比以一行命令来的更优雅呢?
神奇的Awk
不止提取某列这么简单
Awk应该是功能最强大的命令行工具了,厉害的大牛一般不需要编程写脚本,awk就是他们的脚本,而且一般都是单行完成,我们没那么厉害,先从基础学起咯,一点点来
awk中一般数字就代表列号(除了0,因为0表示打印整行)
想输出第4列:awk '{print $4} a.gtf' ,这样会直接输出到屏幕,当然可以直接将结果保存到一个文件中,这个操作叫做重定向awk '{print $4} a.gtf' >a.txt ,又或者可以利用管道符号将结果进行下一步操作awk '{print $4} a.gtf' | COMMAND
Awk本身就是一门编程语言,因此也要有输入文件、命令、输出文件。需要注意的是,awk的全部需求都是要写在{}中,并且是对文件一行一行进行操作。
默认情况下,awk会把文件的每一行按照空格分成几部分(Filed),当然按什么分隔可以人为设定,然后将每一个部分赋给$1, $2,... $N这样的变量【$N表示当前行的最后一部分,另外前面也提到了,$0表示输出整行】,然后默认的操作是print
实际操作
其中有许多实用小操作哦
#下载测试数据
wget https://raw.github.com/nachocab/nachocab.github.io/master/assets/transcriptome.gtf
下载好一个文件后,首先你肯定要看看它包含了什么,于是可能你会用cat或者head查看,保证让你看过就想放弃生信~
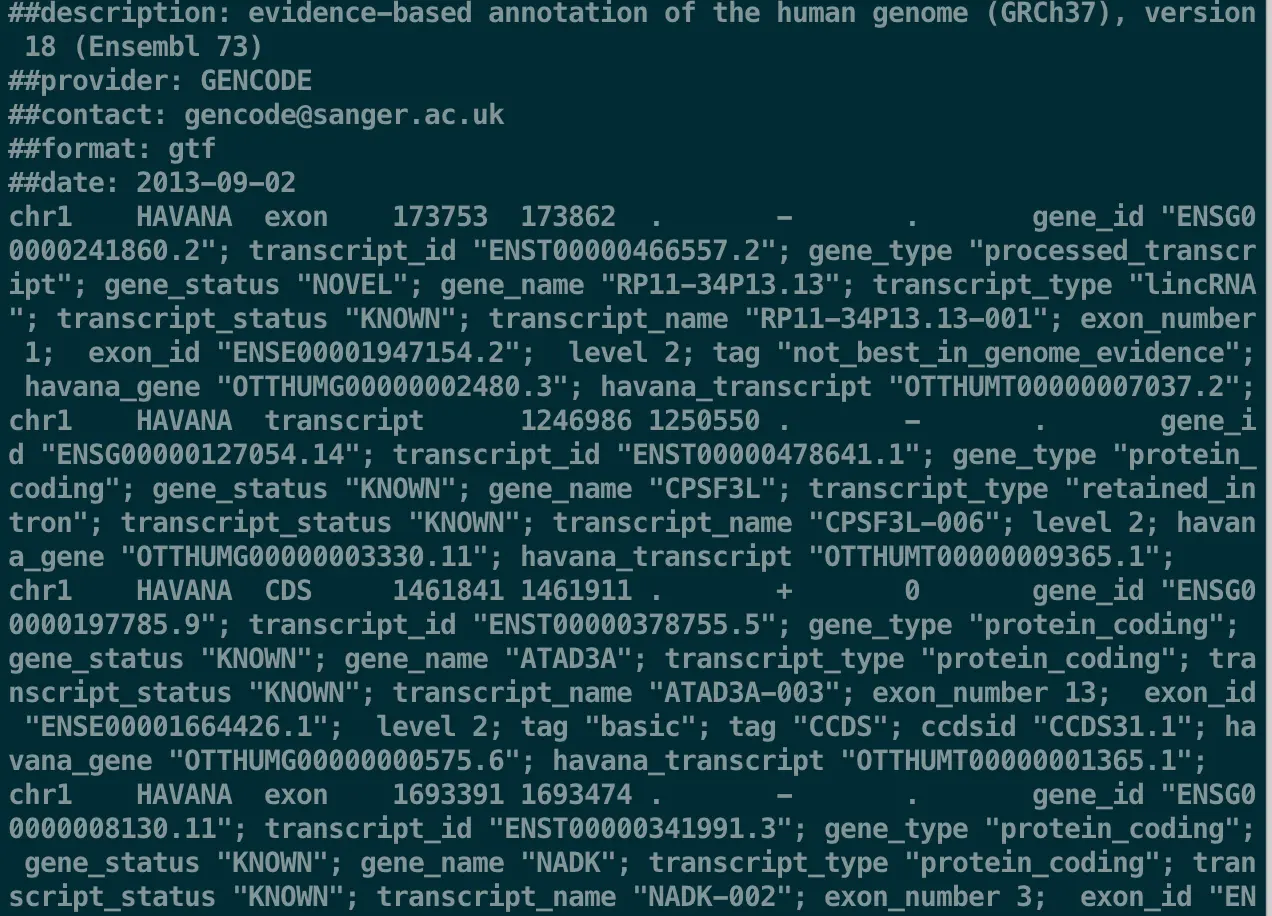
但任何时候,一旦有放弃的念头,就要想想,世界上这么多人做相似的事情,为什么他们没有放弃?是不是有更好的解决办法?试着解决而不是逃避,这样会发现越来越有趣
less -SN transcriptome.gtf 再看一下,发现是不是好多了?并且还带上了行号?这就是-SN的作用
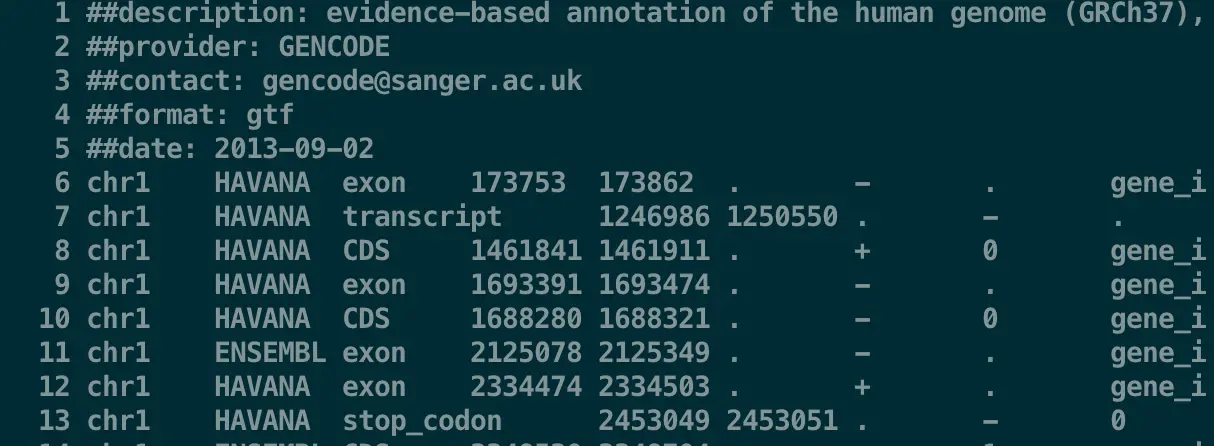
【这样就可以上下翻页了,并且shift + g跳到底部,gg返回顶部,非常好用】
这个GTF文件有9列(虽然最后一列很长,但它相当重要),意思分别是
chromosome, annotation source, feature type, start, end, score, strand, and phase
其中值得一提的是最后一列phase:它以键值对的形式给出,同一键值对的键与值用空格分隔,不同键值对用分号隔开,有一些键(Key)是可选的,一些键是大部分GTF文件中都有的,值(Value)是用引号包围的【这些信息都可能为以后准确提取大量文本提供帮助】
问题
我么知道编码区有的存在许多外显子(Exon),也有的只有一个。那么现在要从GTF中提取只有一个外显子(exon)的蛋白编码基因,该怎么做?
首先我们要找到基因有哪些:
awk '$3 == "gene"' transcriptome.gtf | head | less -SN
# awk所有的规则制定都在单引号''中完成,其中$3指定第三列,后面==表示要满足什么要求,具体的匹配条件需要用双引号包围,例如“gene”
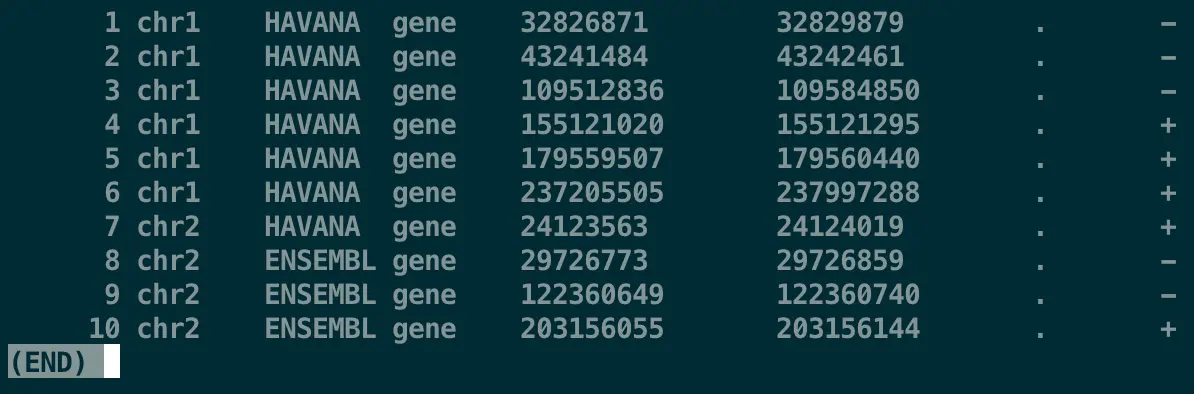
对第3列进行过滤后,我们只想要过滤后的第9列
awk '$3 == "gene" {print $9}' transcriptome.gtf | head | less -S
# 前面提到了,awk需要三大块:输入内容、运行程序、输入内容。如果不写{}的话,即不指定输出什么内容,awk默认输出全部内容
gene_id
gene_id
gene_id
gene_id
...
但是第9列不是很多键值对组成的吗?这列怎么只有一列gene_id呢?
这里又需要学习一下awk对列(column)的理解【有没有发现,生信中许多知识都是交叉学习的,学这个知识点往往能带出来其他知识,逻辑能力就是这样锻炼的吧】
我们知道,awk默认是按照空白进行分隔。空白包括两种:空格和tab,那么awk是按照什么分隔的呢?看个例子就知道:
echo "1 2 3" | awk '{ print $2 }'
# 2
echo "1\t2\t3" | awk '{ print $2 }'
# 结果为空
虽然awk默认用空格(space)分隔,但是我们也可以进行指定:
echo "1,2,3" | awk -F "," '{ print $2 }'
# 设置按,分隔,结果输出2
echo "1 2 3" | awk -F "\t" '{ print $2 }'
# 设置按\t分隔,结果为空
echo -e "1\t2\t3"" | awk -F "\t" '{ print $2 }'
# 设置按\t分隔,结果为2
看到这里是-F起的作用,它可以选择Field。看到第二行命令没有输出结果,因为它找不到任何的\t进行分隔,因此只能将全部内容输出,但全部内容却作为了一整列,我们想输出第二列$2,因此没有内容,但如果命令将$2改为$1,结果就是全部的1 2 3
好了,知道了-F的重要性以后,我们就知道为什么上面的awk '$3 == "gene" {print $9}' transcriptome.gtf 命令只输出了gene_id:没有指定分隔符,对整个文件来讲这9列是tab分隔,而在第9列内部,键与值之间又是空格分隔。因此如果不指定分隔符的话,默认就是找空格所在位置,也就是第九列的起始gene_id处;指定了分隔符以后,就可以按照tab找到真正的第九列
awk -F "\t" '$3 == "gene" { print $9 }' transcriptome.gtf | head | less -S
gene_id "ENSG00000225828.1"; transcript_id "ENSG00000225828.1"; gene_type "protein_coding"; gene_status "KNOWN"; gene_name "FAM229A" ...

刘小泽写于19.3.2
上一次说到了怎样找到特定的列(比如:
$3 == "gene"以及返回特定的列({print $1})。今天继续探究上次的文件
我们看到GTF文件中存在许多的键值对,上一次我们知道了怎么将全部的第九列提取出来:
awk -F '\t' '$3 == "gene" {print $9}' transcriptome.gtf | head | less -S
那么现在,如果想提取第9列中的关于gene_type 信息(也就是第三列),需要在上面代码的基础上用awk再次指定分隔符为; ,接着提取第三列就好
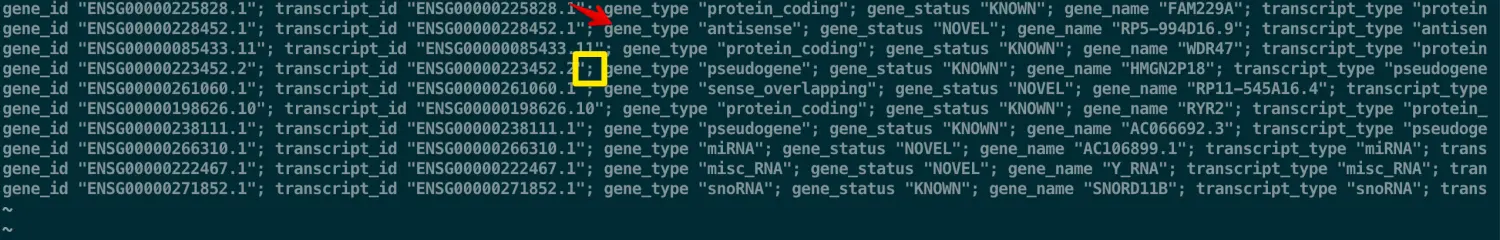
awk -F '\t' '$3 == "gene" {print $9}' transcriptome.gtf |awk -F ';' '{print $3}'| head | less -S
# 得到如下
gene_type "protein_coding"
gene_type "antisense"
gene_type "protein_coding"
gene_type "pseudogene"
gene_type "sense_overlapping"
gene_type "protein_coding"
gene_type "pseudogene"
...
因为存在许多的gene_type,而我们就是想要蛋白编码区(protein_coding)的,于是可以指定筛选条件:【需要注意一点:“protein_coding"的前后引号需要用反斜线进行转义,便于awk识别筛选条件】
awk -F "\t" '$3 == "gene" { print $9 }' transcriptome.gtf | \
awk -F "; " '$3 == "gene_type \"protein_coding\""' | \
head | less -S
或者:如果感觉反斜线的使用不美观或者经常忘记使用,那么也可以用~表示部分匹配,只要搜索的列中包含你输入的字符,那么就会输出结果
awk -F '\t' '$3 == "gene" {print $9}' transcriptome.gtf | \
awk -F ';' '$3 ~ "protein_coding"' |\
head | less -S

到这里我们已经输出了所有的protein_coding基因,那么之前的问题又来了:
如何输出只包含一个exon的protein_coding基因呢?
这里发现:开始我们选择的提取条件是第三列的gene,但是第三列除了gene还有其他信息,统计一下:
awk '{print $3}' transcriptome.gtf | sort | uniq -c | sort -r
# 有用信息如下
1203 exon
730 CDS
273 UTR
228 transcript
94 start_codon
75 stop_codon
63 gene
最多的还是exon,于是就可以将$3 == "gene"改成$3 == "exon" ,挑出全部的exon基因名信息(在分号隔开的第5列)
awk -F '\t' '$3 == "exon" {print $9}' transcriptome.gtf | \
awk -F ';' '$3 ~ "protein_coding"' |\
head | less -S
# 得到的结果
1 gene_name "NADK"
2 gene_name "C1orf86"
3 gene_name "RER1"
4 gene_name "SLC2A5"
5 gene_name "LZIC"
6 gene_name "FBXO6"
7 gene_name "NPPA"
8 gene_name "PLOD1"
9 gene_name "FHAD1"
10 gene_name "CLCNKA"
...
# 还有另外一种实现方式(因为发现GTF第9列中既有分号又有空格分隔,还有引号。因此可以直接将分号和引号去掉,然后直接指定分隔符是空格),接下来就可以直接指定第6列为protein_gene时将第10列name打印出来
awk -F '\t' '$3 == "exon" {print $9}' transcriptome.gtf |\
tr -d ";\"" |awk -F ' ' '$6 == "protein_coding" {print $10}'|\ head |less -S
# 结果如此,更加方便下面进行
NADK
C1orf86
RER1
SLC2A5
LZIC
FBXO6
NPPA
PLOD1
FHAD1
这样就得到了exon与protein_gene的对应关系
接下来到了计算每个基因对应的exon出现多少次的时候了,其实就是统计上面的结果
awk -F '\t' '$3 == "exon" {print $9}' transcriptome.gtf | tr -d ";\"" |awk -F ' ' '$6 == "protein_coding" {print $10}' | sort | uniq -c | sort -r >gene_exon_num.txt
# 结果
4 TTN
4 KIF12
3 LAT2
3 ITSN2
3 CBWD6
2 UBXN11
...
然后看看有多少基因是只有一个exon的:
awk '$2 == 1' gene_exon_num.txt | wc -l
刘小泽写于19.3.9 终于搞完了预答辩,想想昨晚熬夜到四点做完了PPT,真是不容易啊,推送继续来,未来的时间会越来越充裕,我也期待继续和大家一起学习😊
今天继续挑一些我个人认为比较重要的单行命令,记录下来
循环少不了
尤其是在处理多条染色体的时候,比如人类1-22条常染色体+X、Y、M染色体
有几个注意的问题:
# 只处理数字的话,例如只处理常染色体
for i in {1..22};do echo $i;done
# 如果再加上其他非数字,例如这样
for i in {1..22,X,Y,M};do echo $i;done
# 得到的结果是不对的,并不是输出1-22以及XYM,而是输出
1..22
X
Y
M
# 如果想同时得到数字和非数字输出,要把非数字内容放大括号外边,并且空格分隔
for i in {1..22} X Y M;do echo $i;done
# 如果想逆序输出,就在大括号中先写大数,后写小数,例如
for i in {5..2} X Y M;do echo $i;done
5
4
3
2
X
Y
M
用cut提取列
我们都知道的用法是:
# 提取1-5以及10-15列(注意中间的逗号)
cut -f1-5,10-15 data
# 还可以指定分隔符(默认tab分隔)
cut -d' ' -f1-5 data
另外,如果想从第5列输出到最后一列,其实我们不需要知道一共有多少列,直接这样:
cut -f5- data
# 注意5后面的-就是表示一直到最后一列
这样普通的用法cut还是很方便的,但是相比awk,cut还是略显简陋,因为不支持自定义列,例如:
# 先提取第5列,然后提取第2列(注意如果操作gtf文件,需要看看有没有头注释信息,有的话需要先去掉)
cat gencode.v28.gtf | grep -v "#" | awk '{print $5"\t"$2}' | head
# 但是用cut就不能实现
cat gencode.v28.gtf | grep -v "#" | cut -f5,2 | head
# 得到的结果还是按照先第2列后第5列排序
多学点awk
假设现在有一个test.txt文件,其中包含了1-22条染色体的基因信息。其中第一列是基因名,第2列是染色体编号。
我们现在想按照test.txt中不同染色体的信息区分开,1号染色体基因信息存放到chr1.txt中,2号染色体的基因信息存放到chr2.txt中…
可能一开始我们想这么做
for i in {1..22};do awk '$2 == $i' test.txt >chr$i.txt;done
但是要知道awk和shell是不融合的,awk中并不知道在shell中定义的$i
因此我们指定寻找$2 == $i,awk不清楚要找什么,但事实上,我们就是想让awk去根据在shell中设定的变量去引用,只需要加一个参数-v:
for i in {1..22};do awk -v i=$i '$2 == i' test.txt >chr$i.txt;done
-v的作用就是在awk中引入变量variable
例如还有:
# 自定义过滤指标(例如要求第四列值大于10)
for i in {1..22}; do awk -v i=$i -v threshold=10 '$2 == i && $4 > threshold' test.txt > chr$i.txt; done
今天时间有限,先发这些吧
刘小泽写于19.3.10
继续学习单行命令
Case1 - awk
如果有一个文件包含两列,一列是尝试(attempt)次数,一列是成功(success)次数,我们想计算总的成功率,也就是自定义输出结果。
逻辑上很简单,需要知道总的尝试次数和总的成功次数,那么怎样获得这两个值呢?也就是说,我们怎样将第一列累加并赋值给attempt,第二列累加值给success?
# 创建一个新的变量就需要先用BEGIN指定,然后再END中输出
awk 'BEGIN { sum_attempt = sum_success = 0; }{ sum_attempt += $1 ; sum_success += $2} END {print "Attempt: ", sum_attempt, "Success: ", sum_success, "Rate: ", sum_success/sum_attempt*100,"%" }' test.txt
这样结果输出的就会是类似这样:
Attempt: 30
Success: 15
Rate: 50%
如果有一个文件,第一行是头信息,我们想去掉第一行,然后只选出来其中的第4列:
awk 'NR>1 && NF == 4' test.txt
其中NR指的是行号(Number of row),NF指的是列号(Number of field)
Case2 - grep
如果我们要找到以“bioplanet”开头的记录,需要使用^
grep '^bioplanet' test.txt
Case3 - sort
如果有一个文件的第2列是染色体编号,第3列是染色体前起始位置,现在我们想先根据染色体编号排序,再按照起始位置排序,就可以利用sort
sort -k 2n -k 3n chr.txt
-k就是指定按照哪一列进行排序,但是需要注意的是,默认按照ASCII码排序,即100小于11,于是如果要按照真实阿拉伯数字大小排序,需要加上参数-n
Case4 - uniq
如果一个文件中第3列包含许多重复的字符,例如分组信息A、B、C三组,看看每一条记录在哪个分组信息中,最后计算各个分组信息的情况
awk '{print $3}' test.txt | sort | uniq -c
# 这里-c表示count
一般uniq与sort是分不开的,并且是先sort后再uniq
如果现在有两个文件,例如:
$ cat test1.txt
A1BG RXRA
A2M-AS1 GABP
A2M SRF
A4GALT GABP
AAAS CTCF
$ cat test2.txt
ACAT2
A1BG
ACTA1
ACTA2
ADM
AEBP1
我们想找他们的第一列是否有相同的结果
首先就是将这两个文件合并在一起,然后取出第一列,并排序,排序后找共同值【也可以理解成为取两文件某一列的交集 intersection】
$ cat test1.txt test2.txt | awk '{print $1}' | sort | uniq -d >intersection
# 结果就是A1BG
这里看到用了-d的参数,意思就是只输出出现次数大于1次的行
【如果要取两个文件的并集 union,就是去掉它们共同有的成分,直接把uniq的-d参数去掉即可】
$ cat test1.txt test2.txt | awk '{print $1}' | sort | uniq >union
A1BG
A2M
A2M-AS1
A4GALT
AAAS
ACAT2
ACTA1
ACTA2
ADM
AEBP1
如果想找在第一个文件中存在而在第二个文件中不存在的行,需要先找交集(上面已经完成了),然后从第一个文件中剔除交集的部分
$ awk '{print $1}' test1.txt | cat - intersection | sort | uniq -u >test1-only
A2M
A2M-AS1
A4GALT
AAAS
注意到cat 后面多了一个-,意思是将前面awk的标准输出结果存到-中,方便调用;另外这里用了-u的参数,意思是只输出出现一次的结果
Case5 - join
这个用法和R中的merge差不多,就是根据两个文件某一列,将共同的行输出,利用join可以实现,但使用前必须进行sort
# 这里都按第一列进行排序
$ sort -k1 test1 >sort.test1
$ sort -k1 test2 >sort.test2
$ join sort.test1 sort.test2
# 结果就是 A1BG RXRA,可以看到,除了输出了二者第一列的交集A1BG,还输出了后面的内容
join根据第一列进行融合,如果要指定按照其他列,可以join -j
更多情况是:要比较的两个文件列数不一致,前面例子中都是根据第一列,但现实中可能根据第一个文件的第二列,根据第二个文件的第三列,然后找他们的共同之处,并提取出共同的行
# 首先还是要将文件根据不同的列进行sort
$ sort -k2 a1> sort.a1
$ sort -k3 b1> sort.b1
$ join -1 2 -2 3 sort.a1 sort.b1
这里的-1 2 -2 3就是指的利用第一个文件的第2列和第二个文件的第3列
来看一个更加贴近实际的例子:
$ cat >exp.bed
chr1 26 39
chr1 32 47
chr3 11 28
chr1 40 49
chr3 16 27
chr1 9 28
chr2 35 54
chr1 10 19
$ cat >exp_lengths.txt
chr1 58352
chr2 39521
chr3 24859
目标:把长度信息贴在bed文件每一行后面
$ sort -k1 exp.bed >sort.exp.bed
$ sort -k1 exp_lengths.txt >sort.exp_lengths.txt
$ join -1 1 -2 1 sort.exp.bed sort.exp_lengths.txt
chr1 10 19 58352
chr1 26 39 58352
chr1 32 47 58352
chr1 40 49 58352
chr1 9 28 58352
chr2 35 54 39521
chr3 11 28 24859
chr3 16 27 24859