088-忍受不了Blast比对的龟速怎么办?
刘小泽写于19.2.27
将序列比对到nt、nr、uniprot、pfam等数据库是常有的事,blast无疑是最常用的比对软件,但是它的速度一直提不起来,非常耗费时间。估计比对等不到blast的结果了,于是想到了另一款软件Diamond
Diamond简介
最大的特点就是快,比blast快500-20000倍;
对于长序列,支持Frameshift alignments;
占用资源更少,但只是相对blast来讲;
自定义多种输出格式
记得引用:
Buchfink B, Xie C, Huson DH, “Fast and sensitive protein alignment using DIAMOND”, Nature Methods 12, 59-60 (2015). doi:10.1038/nmeth.3176
Diamond使用
还是conda安装好diamond,说明书在此:https://github.com/bbuchfink/diamond/raw/master/diamond_manual.pdf
比对之前先构建好目标数据库的索引
最近下载了一个nr数据库,解压完是110G,命名为nr.faa
diamond makedb --in nr.faa -d nr -p 20
结果会生成一个nr.dmnd的索引文件
我用了20核构建了110G的nr数据库,用时5726.56s,作为参考
Pfam数据库7.1G,24核用时190.931s,还是非常快的
附带数据库下载地址:
Nr:http://mirrors.vbi.vt.edu/mirrors/ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz
Pfam:ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam32.0/Pfam-A.fasta.gz
然后可以进行比对了
# 因为要将核酸序列比对到蛋白库,因此使用blastx功能
diamond blastx -d nr -q reads.fna -o nr.matches
默认输出格式是BLAST的tab结果,-o指定输出文件名
需要注意的是:
这个过程需要消耗大量的内存与硬盘(用于存放临时文件),如果资源不够,可以设置
-b参数调整默认的比对模式(fast模式)是针对短序列的,如果要比对的序列比较长,建议使用sensitive模式,设置
—sensitive或者--more-sensitive运行时间不与比对文件大小成正比,相反,文件越大(比如大于1M reads)比对速度越快
比对结果中的X表示Masked residues
默认的阈值是0.001而Blast是10,相比之下,diamond比对更严格,得到的weak hits会更少
比对结果进行过滤
一般运行的结果都会比较大(上百兆文件),但其实nr.matches这个文件是冗余的。转录本毕竟也是软件预测拼接得到的,因此预测出来的多数转录本是没有意义的,并不能认为全部都是有效转录本,那么对于这个比对结果,我们需要过滤一下,然后再检测物种相似性
首先对比对序列的长度从长到短排序
cat nr.matches | sort -n -r -k4 > nr_matches.sort
然后利用Python的pandas
# 脚本来自https://www.cnblogs.com/leezx/p/8603166.html
import pandas as pd
infile = "extract_no_N_200.fasta.diamond.nr.sort"
#infile = "test.sort"
df = pd.read_csv(infile, sep="\t", header=None)
df.columns = ["l1","l2","l3","l4","l5","l6","l7","l8","l9","l10","l11","l12"]
df1 = df.sort_values(['l4'],ascending=False)
df2 = df1.drop_duplicates(subset=[df1.columns[1]], keep = 'first')
df2.to_csv("rm_dup_protein_"+infile, sep="\t", index=False, header=False)
df3 = df2.sort_values(['l3'],ascending=False)
df4 = df3.drop_duplicates(subset=[df3.columns[0]], keep = 'first')
df4.to_csv("unique_protein_"+infile, sep="\t", index=False, header=False)
结果文件如下:可以看到unique仅占原始的的24分之一
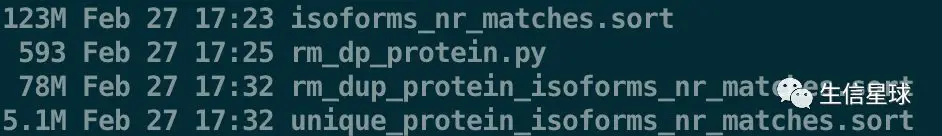
文件内容大致是这样:
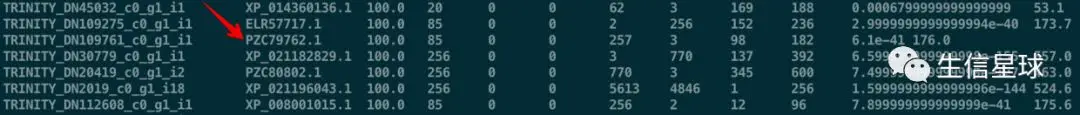
有了非冗余的转录本序列后,我们可以利用第二列信息,也就是Genbank 的ID号继续做Nr注释,看看不同的转录本比对到什么物种