087-问题:转录本ID转换?
刘小泽写于19.2.25
好久没写咯,最近一直在写毕业论文,就给自己一周的时间写完,然后开始学习新知识😎期待!到时不断更的我又会出现
重回正题~Trinity确实是个非常好的软件,官方文档写的十分详细,而且主流的无参转录组分析都推荐使用这款,它可以预测出更长的亚型、更多的基因和转录本,另外它还有几十个小工具,比如不会做热图,不会做火山图都可以借助它来进行可视化,完全不需要本人需要会R知识,直接一个shell命令搞定。去年2月我刚开始接触生信不久,就做了这个内容,因此记得特别清楚那种感觉——不明不白整出来一个自己看不懂的pdf,导出以后,哇这么高大上!
但随着时间的推移,知识是不断更迭的,现在如果还是用老一套程序自带的小工具,就有点落伍了
最近就遇到这样的一个问题
不想用Trinity自带的作图脚本,因为做出来有点nan kan🤥并且不能按照我们自己的需求去定制,这一点是综合性工具的通病。
例如:自带的火山图【想改颜色?想加基因名?想加辅助线?对不起,Trinity不可以】
因此,如果对R语言还算熟悉的话,直接可以从定量结束后,自己去做,无论用limma、deseq、edger,只要你喜欢,都可以实现,并且有太多的可视化包,比如ComplexHeatmap(https://github.com/jokergoo/ComplexHeatmap)做出来的图都很好看。这里为了快速演示学习,我就用了简单的pheatmap功能
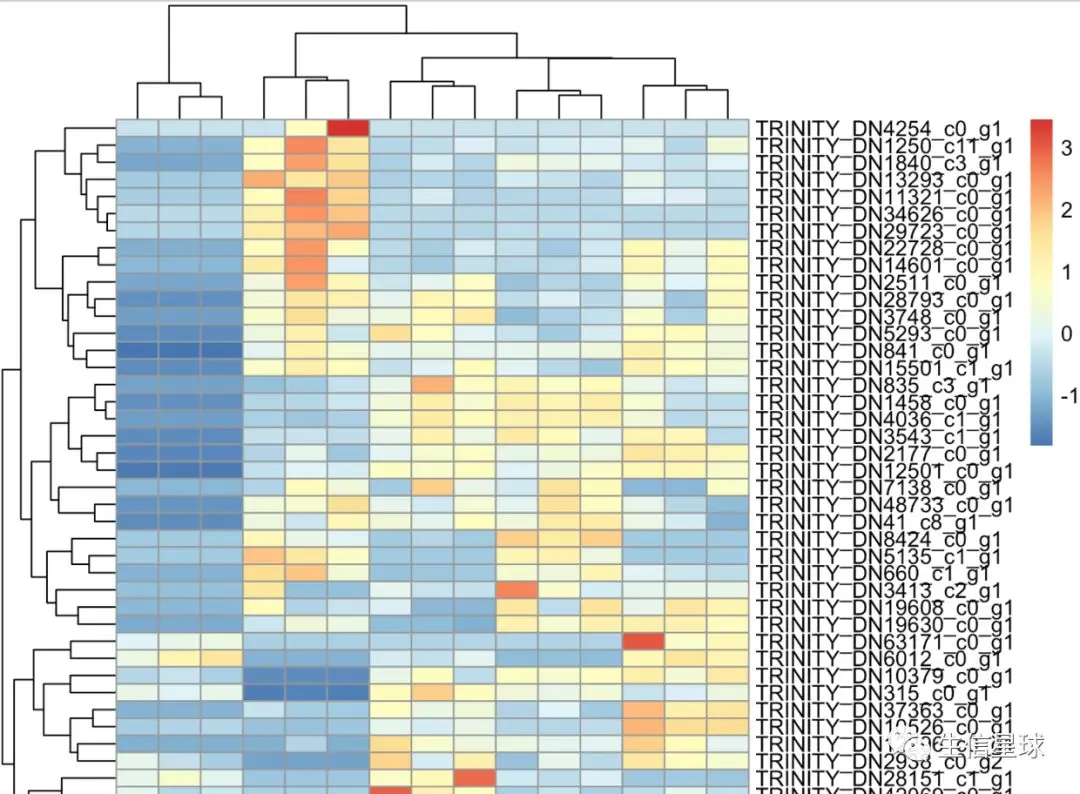
可以看到,我这里截取了前30基因进行不同样本间的热图绘制,结果看右侧全是Trinity,我们自己可能知道是Trinity拼接的转录本(或者也勉强可以叫基因),但是对于别人不清楚Trinity的,就不知道我们做了啥。
别人想看到的,可能仅仅是几十个基因名
例如,在利用DESeq2得到差异基因后,几十个Trinity ID,如何让它们变成基因名,方便下游的功能注释【因为使用clusterProfiler对非模式生物进行注释,需要ID的转换,并且转换列表里肯定没有Trinity ID选项】
肯定不能让我们自己一条一条去找序列然后blast吧~想想都不行!
于是,我们之前对Trinity.fasta做的各种注释派上了用场:nr注释、uniprot注释,其中都有基因信息啊
再说一个经验:不要想着一次就实现批处理,先对一条序列或一个基因做通了,批处理也不再是难事
举个例子:我现在在热图中看到20条基因在样本间差异表达还比较明显,我想想看看它们,先从中抽出来一条让我们写写脚本练练手,就拿TRINITY_DN1458_c0_g1_i1来说吧
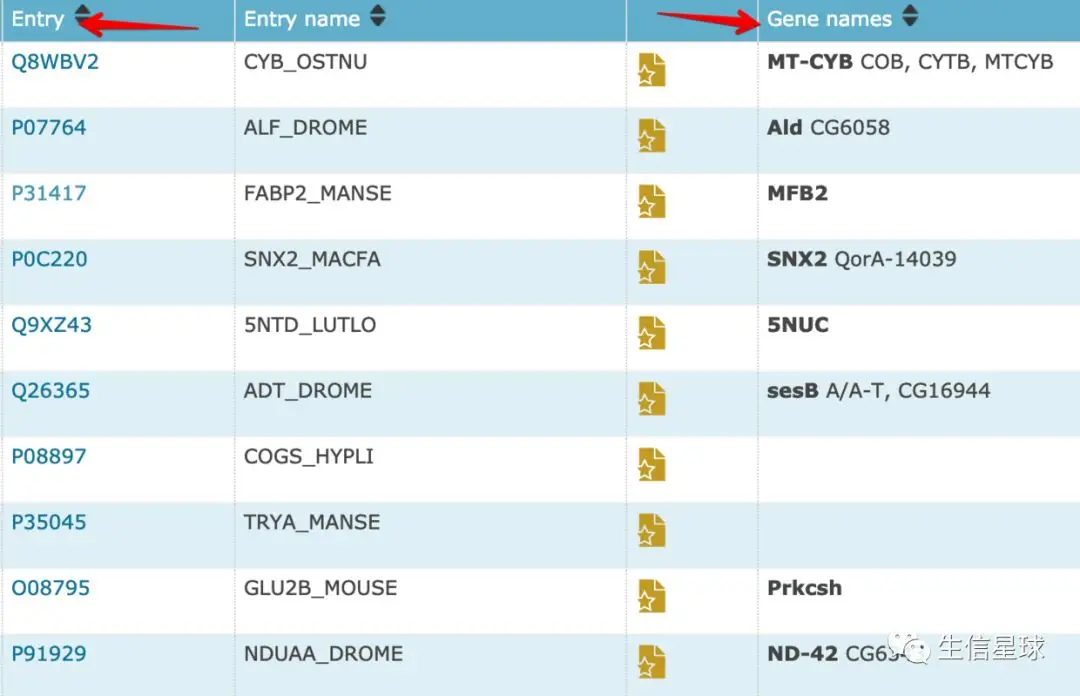
我们之前跑完了Uniprot的注释,结果文件是blastx.outfmt6,重点关注第二列
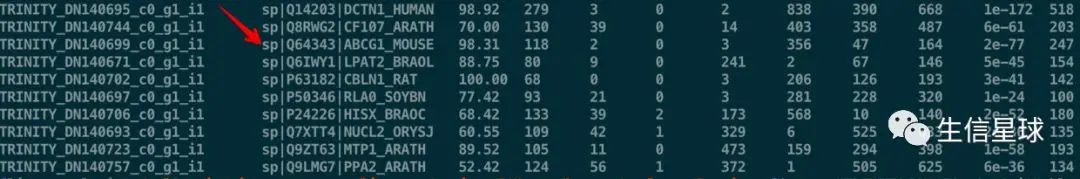
第二列记录了数据库比对到的Uniprot编号与物种,以第一行为例,Q14203是UniProt编号,可以直接查询;DCTN1_HUMAN是比对到的名称和物种
我们看看TRINITY_DN1458_c0_g1_i1这个转录本对应的信息是什么吧:
grep "TRINITY_DN1458_c0_g1_i1" blastx.outfmt6
# TRINITY_DN1458_c0_g1_i1 sp|P31417|FABP2_MANSE 54.89 133 59 1 63 461 1 132 1e-45 153
然后我们需要导出Uniprot ID号
grep "TRINITY_DN1458_c0_g1_i1" blastx.outfmt6 | cut -f 2 | cut -d "|" -f2
# P31417
这样一条基因的就搞定啦!
看看clusterProfiler支不支持吧~
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db)
## [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
## [5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
## [9] "EVIDENCEALL" "GENENAME" "GO" "GOALL"
## [13] "IPI" "MAP" "OMIM" "ONTOLOGY"
## [17] "ONTOLOGYALL" "PATH" "PFAM" "PMID"
## [21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
## [25] "UNIGENE" "UNIPROT"
看到了"UNIPROT"的选项,于是可以愉快地使用bitr函数去转换啦
只不过需要使用自己研究物种的orgdb
或者,使用Uniprot在线工具也行(https://www.uniprot.org/uploadlists/)
如果有多条转录本呢?
一条的会了,多条的就是一个批处理而已
还是首先从Rstudio中导出那些需要转换的,首先这是一个字符型向量,怎么导出为txt呢?由于好久不接触R,有点生疏了😅,问了花花,2行代码搞定【最后花花还附加了一句:一切能放到R里的问题都难不倒我】
top30 <- as.data.frame(top30_gene)
write.table(top30, file="test.txt", row.names = F, quote = F)
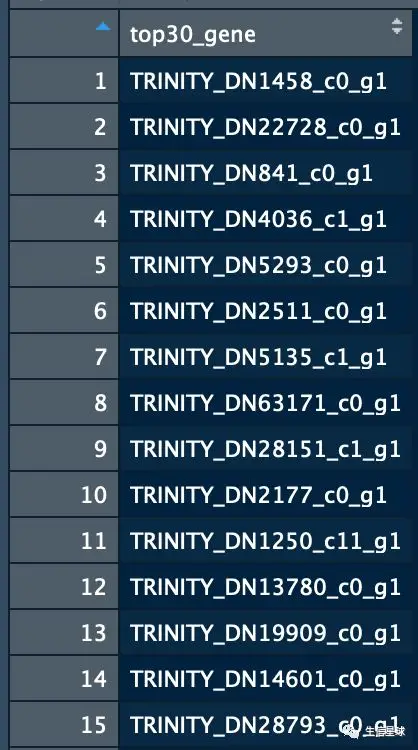
然后,去linux中放到一个文本
# 这里截取一部分
cat >test.txt
TRINITY_DN1458_c0_g1
TRINITY_DN22728_c0_g1
TRINITY_DN841_c0_g1
TRINITY_DN4036_c1_g1
TRINITY_DN5293_c0_g1
TRINITY_DN2511_c0_g1
TRINITY_DN5135_c1_g1
TRINITY_DN63171_c0_g1
TRINITY_DN28151_c1_g1
TRINITY_DN2177_c0_g1
TRINITY_DN1250_c11_g1
然后批处理
需要考虑到有的转录本没有对应的注释,此时就需要指明,否则顺序就乱了;另外还有一种情况需要注意,就是一个转录本存在好几个注释,如下图
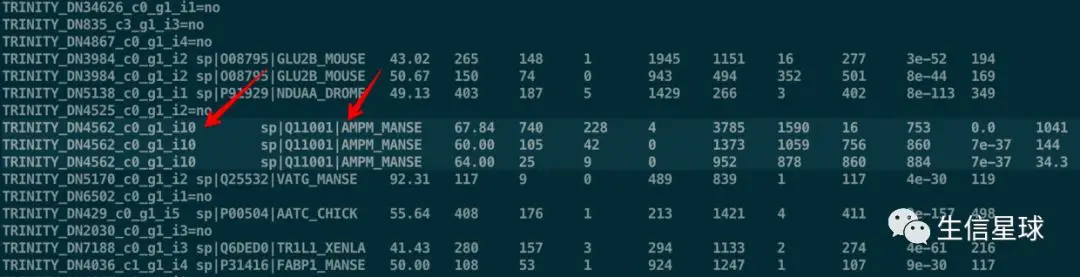
cat top30.txt | while read id;do if grep -q -w $id blastx.outfmt6 ;then (i=${id};grep -w ${i} blastx.outfmt6|cut -f 2 | cut -d "|" -f2);else echo "${id}";fi ;done
cat trans.txt
# 截取一部分
P31417
P31416
P31416
P31416
Q26365
O61387
P0C220
P0C220
Q9CWK8
Q9CWK8
Q2TBW7
当然,这只是最粗略的方法,还有一些问题需要解决,比如多个比对选分值最高;完全相同的比对需要去重复
最后得到了对应的基因名,可以看到,虽然有的转录本可以转换成Uniprot ID,但依然存在对应不到基因的情况,这种在后续注释中可能就要抛弃了