083-基因家族聚类之OrthoFinder
刘小泽写于19.2.11
这是干啥的?
做进化、基因家族分析、比较基因组使用
OrthoFinder is simple to use and all you need to run it is a set of protein sequence files (one per species) in FASTA format.
最新版的是version2, Emms, D.M. and Kelly, S. (2018) OrthoFinder2: fast and accurate phylogenomic orthology analysis from gene sequences. bioRxiv
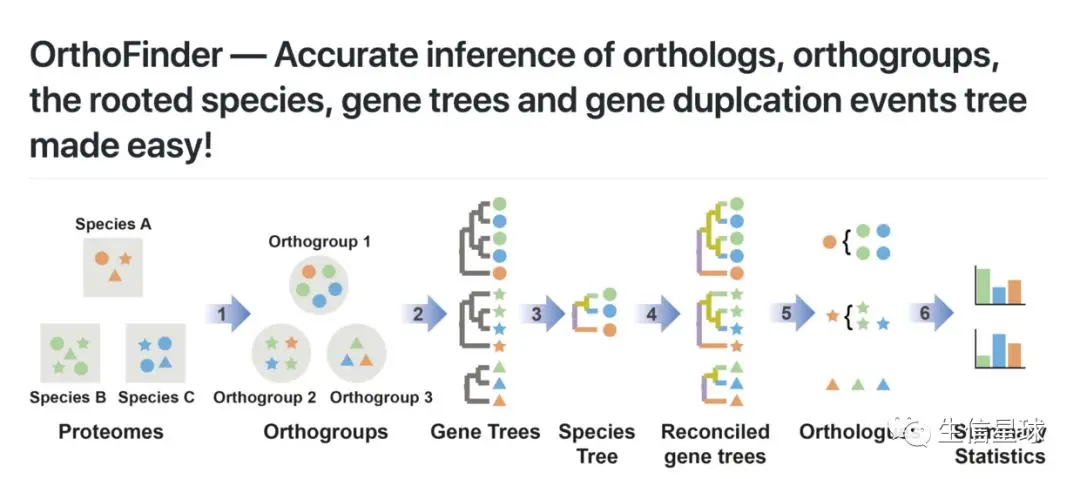
Orthologs: pairs of genes that descended from a single gene in the last common ancestor (LCA) of two species
Orthogroup:(extension of the concept of orthology)the group of genes descended from a single gene in the LCA of a group of species
【all the genes in an orthogroup started out with the same sequence and function】
使用
安装
https://github.com/davidemms/OrthoFinder
需要python2环境
conda install -n orthofinder python=2 orthofinder
source activate orthofinder
软件运行依赖于diamond或者mmseqs2、blast、mcl、fastme
参数设置
orthofinder -f data \ #存放蛋白的fa数据
-S diamond \ #比对模式:diamond,blast,mmseqs,blast_gz
-M msa \ #基因树推断法:dendroblast,msa(推荐)
-T fasttree \ #建树软件:iqtree, fasttree, raxml(推荐),raxml
-t 5 #线程
可以用软件自带的参考数据测试:https://github.com/davidemms/OrthoFinder/tree/master/orthofinder/ExampleDataset
运次过程
因为是小测试数据,因此运行的过程都可以监测:
1. Checking required programs are installed
2. Dividing up work for BLAST for parallel processing
3. Running diamond all-versus-all
4. Running OrthoFinder algorithm
5. Writing orthogroups to file
6. Analysing Orthogroups
7. Best outgroup(s) for species tree
8. Multiple potential species tree roots were identified, only one will be analyed.
9. Reconciling gene trees and species tree
10. Writing results files
结果生成这些文件,存放在Result目录中
Orthogroups.GeneCount.csv
Orthogroups.txt Orthogroups_UnassignedGenes.csv
SingleCopyOrthogroups.txt
Statistics_PerSpecies.csv
Orthogroups.csv
Orthogroups_SpeciesOverlaps.csv
Orthologues_Feb11
Statistics_Overall.csv WorkingDirectory
其中Orthogroups.GeneCount.csv 中每一行代表一个基因家族,每一列表示每个物种的基因家族包含多少基因,比如OG0000000这个基因家族,在1物种中没有,在2物种有1个基因,在3物种有8个基因
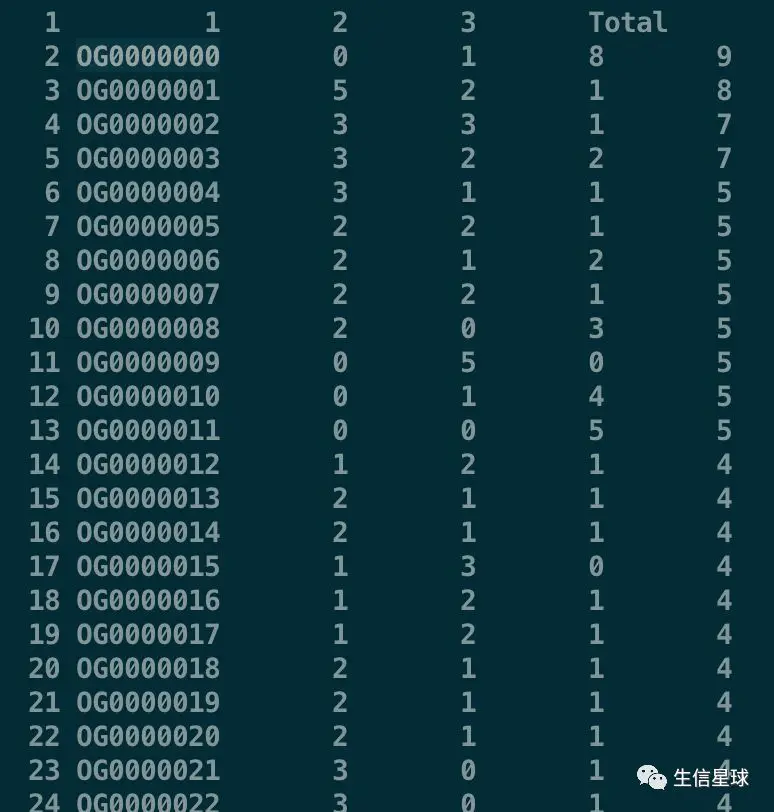
我们选出各个物种中基因数大于0的基因家族,首先看物种1
我们不要第一行,然后看物种1,也就是$2,选出大于0的,然后我们需要的是基因家族编号,也即是第一列
sed '1d' Orthogroups.GeneCount.csv |awk '$2 >0 {print $1}' >1.txt
同理可以看其他物种的,根据这个可以做韦恩图(在线工具、R都可以),看看有哪些是共有的