068-短序列比对了解一下?
刘小泽写于18.12.17
组学分析离不开比对环节,可能流程已经会了,但这不是根本。我们需要再深入学习一下关于短序列比对的一些知识,这样以后再分析的时候,就更能明白我们自己在做些什么
短序列是什么?
一代测序一般都测的比较长(1000bp左右)并且准确率很高,能到99.9%,但最大的缺点就是:这么好的技术却不能大批量应用,只能一条一条测,耗时耗钱。从2005年左右,二代测序的短读长、高通量开始迅速发展,产生的reads和一代有着显著的不同:序列长度明显降下来了(50-300bp),一次测序产量明显上去了(一次上亿条reads)。这里不讨论测序仪怎么做到的输出短序列,只需要知道二代测序带给我们的结果一是短,二是多就行了。
有了输出的短序列,然后呢?
它们的用处一般有两个:一个是比对到参考基因组(也就是重测序 re-sequencing流程),另一个就是相互比对进行叠加(也就是组装的过程,reads=》contig=》scaffold等)。这些工作都是靠比对工具完成的,我们称之为:“short read aligners” OR “short read mappers”
比对工具怎么完成的?
首先,让我们感谢那些**短序列比对工具(以下简称“工具”)**的作者,生信界有了他们才有了不同组合的流程,才推动了不同的组学研究。他们设计的一个好的工具可以在1秒内比对超过一万条序列到人类基因组的30亿bp碱基上
比对一般需要寻找exact matches、missing locations 以及partial and alternative matches/overlaps ,但是这几个任务每个软件都有自己的擅长与不足。另外,根据数据类型(domain-specific)表现也不同
当然,工具也有自己的劣势,大部分的工具只能根据阈值去寻找和目标区域相似的比对结果,高相似性优先,因此有部分会因阈值设定被抛弃;当序列过长或过短时,就会受到限制,例如研究small RNA这种低于30nt的序列,可选的比对工具就是少之又少,因为对比对算法的精确度要求更高
alignment vs mapping
说起比对,我们一般都会想起来这两个词,但是严格来说,它们还是有区别的:mapping强调的是不要求比对的准确度,能有比对就可以,是region层面上的,它的目的就是看看基因组上有没有一块区域可以安放这条序列;而alignment强调精确比对,是碱基层面上的
李恒关于这二者做过自己的解释:
Mapping
- A mapping is a region where a read sequence is placed.
- A mapping is regarded to be correct if it overlaps the true region.
Alignment
- An alignment is the detailed placement of each base in a read.
- An alignment is regarded to be correct if each base is placed correctly.
例如:如果研究SNPs和基因组内变异,应该就是用alignment的工具;如果研究RNA-seq就是用mapping的工具,因为RNA-seq一般我们就想知道这个序列从哪里来的就好了,没必要知道比对上多少
这么多工具,如何选择?
其实工具很多,能被大众接受的很少,因此软件开发之路十分艰难,其实大部分的开发者并没有得到关注。
这里用的最多的当属bwa和bowtie2 ,选择的话一般就是根据喜好和信仰充值了。我们知道bwa是曾经的博德研究所的李恒开发的,国人的肯定要支持;另外华大也开发了一个novoalign 喜欢BGI的也可以关注。但是不管怎样,一个比对项目,可以多用几种工具分析分析,增加自己结果的可信度
另外,在选择工具的时候,我们要根据自己的实际需求,自己做一个心理暗示(mental checklist):
- 比对算法:
global, local or semi-local - 工具遇到INDEL怎么处理?
- 工具可以跨大片段区域比对吗?
- 工具可以进行根据阈值过滤吗?阈值设置?
- 工具可以找到嵌合体比对吗?
选择一个最常用的bwa研究下
bwa的背景
bwa是Burrows-Wheelers Aligner的缩写,应该是目前使用最为广泛的,后期会搭配同作者的samtools使用
当时李恒在发表这个bwa算法的时候,那时已经好多人在用了,还被拒稿,为此他写过一封信:
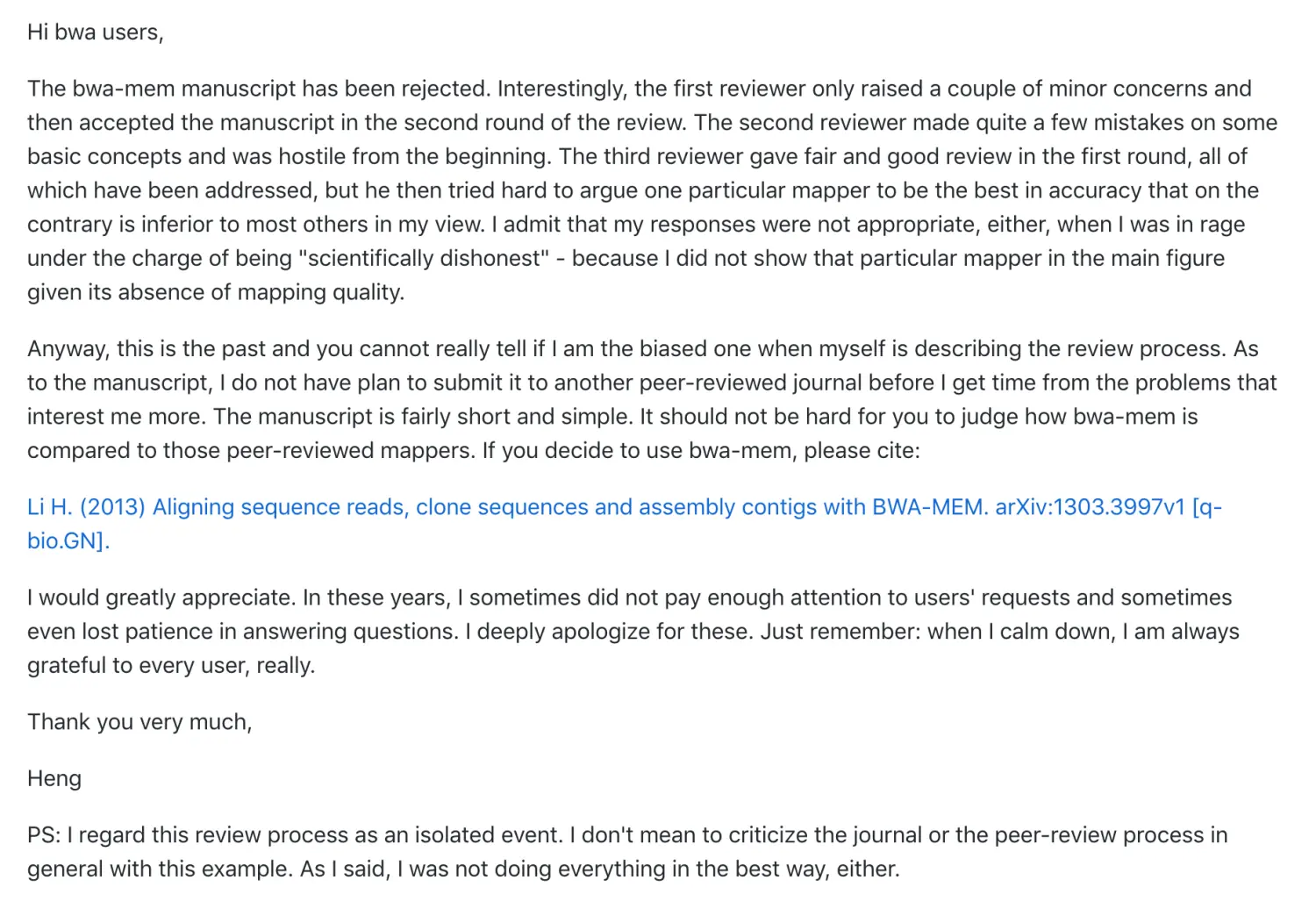
生信有一个特点,就是:更新速度快,可能有的软件还不知道名字就已经被淘汰了。身处前端的bwa也是不断的更替版本,之前的算法是 bwa aln,现在是bwa mem【这里的mem指的是Maximally Exact Matches】。因此如果采用不同的算法,虽然都是bwa,但结果还是有差别。这就是有时让人纠结的地方,因为不清楚作者软件的版本更替,于是,最好的办法就是用熟练github,自己去找(当然,我也在努力的道路上)
bwa准备工作
一般来讲,使用bwa比对之前需要根据参考基因组构建索引index ,目的就是快速搜索基因组。一般参考基因组放一个目录,然后构建的index放另一个目录,随时取用。
bwa小练习
将埃博拉病毒测序的reads比对到它的参考基因组上(命名为1976.fa)
先构建索引
mkdir -p ~/tmp/bwa_test/ref
# 下载基因组(19Kb)[安装entrez-direct]
conda install -c bioconda entrez-direct
efetch -db=nuccore -format=fasta -id=AF086833 > ~/tmp/bwa_test/ref/1976.fa
# 构建索引
ref=~/tmp/bwa_test/ref/1976.fa
bwa index $ref
# ls ~/tmp/bwa_test/ref
然后下载测试数据
mkdir -p ~/tmp/bwa_test/raw && cd ~/tmp/bwa_test/raw
# 获取全部的埃博拉病毒项目的测序数据
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.csv
# 挑选SRR1972739下载
cat runinfo.csv| grep SRR1972739 | cut -d "," -f 10 | xargs -n1 wget {}
# 解压成fq文件 [为了测试,选取前1万条reads]
fastq-dump -X10000 --split-files SRR1972739
比对
比对默认得到sam格式(Sequence Alignment Map),其中包括了所有的比对信息,之后再也不用看fastq了
mkdir -p ~/tmp/bwa_test/align && cd ~/tmp/bwa_test/align
r1=~/tmp/bwa_test/raw/SRR1972739_1.fastq
r2=~/tmp/bwa_test/raw/SRR1972739_2.fastq
ref=~/tmp/bwa_test/ref/1976.fa
baw mem $ref $r1 $r2 > bwa.sam

至于Bowtie2,也有自己的用法,不过是将索引换成了
bowtie2-build, 然后比对的时候bowtie2 -x $ref -1 $r1 -2 $r2 > bowtie.sam运行结束后,会给个报告

软件的比较?
如果只考虑时间的话,bwa要快一些,当然这也是有原因的:bowtie2产生的结果中包含有更多的信息,例如,同时看下它们的tag部分:
cat bwa.sam | cut -f 12-20 | head

cat bowtie.sam | cut -f 12-20 | head
