076-为什么要学编程?
刘小泽写于19.1.8-1.10 这部分内容学自《Perl在生物信息学中应用》
其实之前我也一直困惑这个问题?“为什么一定要自己学习编程,那么多写好的工具,直接拿来用不是更方便吗?我们毕竟是攻城狮而不是程序猿。”我如是安慰自己。
但所有的事情都有一样的规律,现在认为不重要,只是因为还没到那个程度。的确,开始我也想学编程python、perl的书也准备了,但却一直用不上知识,于是就产生了间断期。最近,有强烈的需求想要自己去探索数据的时候,却有点摸不着头脑了,因为现成的工具并不能满足我的全部需求,而且大脑里已经想象出了怎么去一步步处理,就是做不出来。这一次,我真的好想学习一下编程技术,不要求造多好的轮子,只要求自己用到的时候,有想法,能实现,就够了。之后的事情,就要等到再进阶,再有需求的情况去做了。
Perl和Python之间,我先选择Perl
这二者都很好,但是精力有限,只想先看懂简单的代码流程,另外perl one-line命令一直吸引着我,想借此机会探究一下,伊现富老师(https://github.com/Yixf-Education)翻译过一本Perl的书籍《Perl语言在生物信息学中的应用—基础篇》。三天的时间看完了这本书,记录下个人认为重点的一些地方,里面有许多知识需要不断揣摩
什么是编程语言?
指定一系列语法规则指导计算机运行,编写的程序叫源代码,需要翻译成机器语言才能让计算机识别运行。机器语言是二进制,难以肉眼读取编写,因此还需要解释器(编译器)才能运行某种编程语言写的程序(如perl解释器运行perl程序)
编程过程
例如:我们手头有一个DNA序列想看下其中的调控元件数目,大脑中的想法框架应该这样构建:
确定输入–新的DNA序列
问题:输入文件名=》文件是否存在=》打开文件
确定查询—调控元件列表
就像DNA序列一样也要添加进来
进行查找–算法
一般会有好几种解决办法:
算法一:读入一个调控元件,从头到尾对DNA查找,然后进行下一个调控元件;
算法二:一次性读取DNA序列,再在DNA的每一个位置对调控元件进行查找
输出
伪代码帮记录
灵感最重要,先将整体思路用语言先写出来。还是上面例子,我们可以这样写
get the name of DNA file
read in the DNA file
for each regulatory element
if element in DNA, then
add one to the count
print count
之后伪代码可以变成注释信息,方便自己一步步去写需要的代码,并且方便交流
常用元素
命令解释
第一行#起始:
#! /usr/bin/perl -w,告诉计算机这个是perl程序。-w表示遇到错误发出警告语句
分号结尾,如赋值语句
$DNA='ATCGCTACG';,将右边的真实DNA信息存储在perl程序中的DNA变量中变量
只由大小写字母、数字、下划线组成,不能数字开头
$开头的叫标量变量,储存字符串或各种数字,但是一个标量一次只能存储一种类型【不能以数字开头;变量名大小写敏感】换行
print $DNA \n\n表示输出DNA后中间空一行再打印其他单引号与双引号
单引号属于强引用,其中的任何东西都会输出为字符,例如:
"\n"会打印输出换行符,'\n'只会打印\n文件句柄
就是处理文件需要用的东西,例如
$protein=<PROTEIN>中的PROTEIN大小写没有规定,但一般使用大写表示;
其中尖括号表示输入操作符,就是利用这个
<PROTEIN>从外部读入数据,一般操作是:$proteinfilename = 'NM_021964fragment.pep'; open( PROTEINFILE, $proteinfilename ); # 然后就可以把读入的PROTEINFILE存到数组中常用的句柄还有:
STDIN(从键盘上读取用户的输入);STDOUT把输出内容输出到屏幕(默认)或到其他位置;错误信息定向到STDERR,例如:print STDERR "Bad info\n";pop数组末尾拿掉一个元素;shift开头拿掉一个元素;unshift添加一个元素到数组开头;push添加一个元素到数组结尾Perl中有三种主要数据类型:标量变量、数组和散列/哈希(关联数组)
数组
@开头,是储存多个标量的变量@bases = ('A', 'C', 'G', 'T'); print @bases; #输出结果没空格 print "@bases"; #输出结果有空格$dna[2]表示对数组@dna取第3个元素(因为起始索引为0)数组可以用
=拷贝:@out = @in$count = @input得到的标量变量count是数组input中元素数目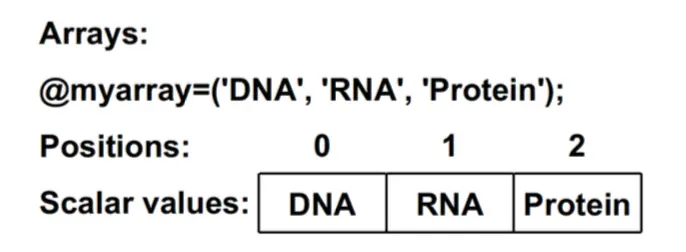
散列/哈希
散列以
%起始,如my %dictionary = (...);。取元素时,散列和数组一样用$,因为查找返回的都是标量值,散列与数组不同之处就是:散列用{},数组用[]获取散列的所有键构成的数组:
@keys = keys %my_hash;;获取散列所有值构成的数组:@values = values %my_hash;利用键值对可以快速提取信息,例如基因名作为键,基因相关信息作为值【注意:不能把正则表达式或字符集作为散列的键,必须是简单的标量值,比如一个字符串或一个数字】对散列进行排序:
@sorted_keys = sort keys %my_hash;
括号:在复杂的运算中指明运算的顺序,如:
(2+3)/3,不加括号就先运行3/3
有用的操作
连接
- 使用
.,$DNA3=$DNA1.$DNA2,然后print "$DNA3\n"; $DNA="$DNA1DNA2",然后print "$DNA3\n";print $DNA1, $DNA2, "\n";或print "$DNA1$DNA2\n";$protein=join('',@protein),需要指定一个字符串,会放在数组各个元素之间,当分隔字符串为空字符串时,就将各个元素连起来
替换–s///
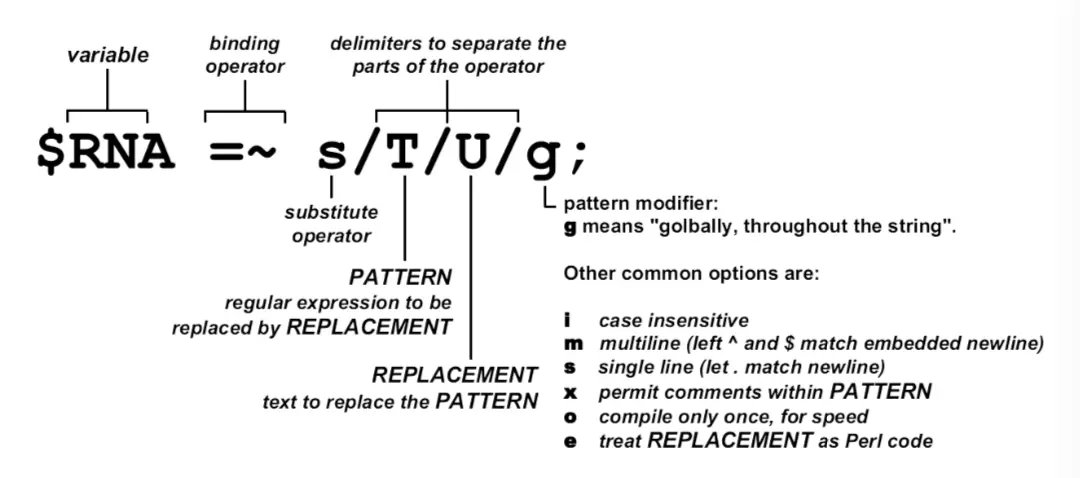
注意到=~,它是绑定操作符,就是将右边的操作应用到左边变量的字符串上;
然后s/T/U/g,s表示替换(substitution),第一个斜线后跟着T,表示字符串中被替换的元素;第二个斜线后跟着U,是要替换成的元素;g表示全局
因此,这句命令的意思就是:把$RNA变量存储的字符串数据中所有T变成U
另外,如果要删除换行符或者空格或制表符,使用$RNA =~ s/\s//g
反转数组–reverse
reverse @bases ,例如:
@bases = ('A', 'C', 'G', 'T');
@reverse = reverse @bases;
print "@reverse\n\n";
# T G C A
互补–tr
tr 其实也是替换的作用,但是对于求序列的互补比较方便,第一个斜线后是要被替换的内容;第二个斜线后是替换后的内容,且位置对应进行替换
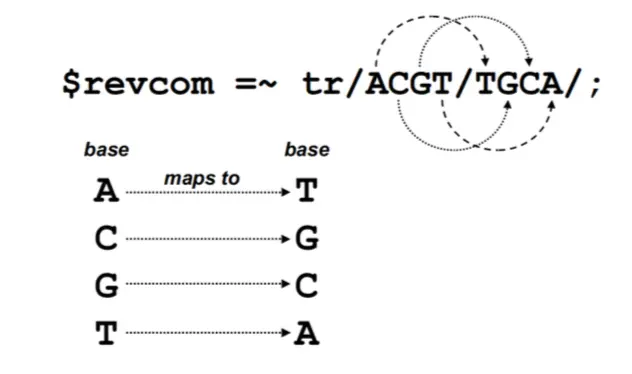
因为tr函数会返回在字符串中找到特定字符的数目,而且即使替换字符集为空,原始字符集也不会改变,因此还可以作为计数器,如:$base = ($dna =~ tr/ACGTacgt//);就可以统计所有碱基数
获取长度–scalar
scalar @bases ,对于上面的例子,得到的结果就是4
任意位置插入–splice
splice ,例如:
@bases = ('A', 'C', 'G', 'T');
splice ( @bases, 2, 0, 'X' );
print "@bases\n";
# A C X G T
参考:http://blog.sina.com.cn/s/blog_4d3a41f40100m29m.html
拆解–split
@DNA=split('',$DNA) 将$DNA拆解成单个字符的@DNA数组【与join相反】
取子集–substr
$base = substr($DNA, $position,1)
变量初始化
声明变量后需要赋一个值,如果不进行初始化,它的值将被定义为'undef’,对于undef,在数字操作中表示0,在字符串操作中表示空字符串
循环
foreach循环【对一个数组中的元素进行循环处理时比较方便】
# 统计碱基数目 foreach $base (@DNA){ $count++; #效果等同于$count += 1 }当然,如果不指定从数组中读取的标量变量,如
foreach (@DNA),perl会使用特殊变量$_while循环
while ($DNA =~ /a/ig) {$a++} #统计A碱基的数目(i表示不区分大小写)for循环示例
# DNA翻译成蛋白序列(codon2aa是一个子程序,记录了密码子与氨基酸的对应) for (my $i=0; $i<(length($dna)-2);$i += 3){ $protein .= condon2aa (substr($dna, $i, 3)); } return $protein;其他一些常用格式
while(CONDITION) {BLOCK} until(CONDITION) {BLOCK} for(INITIALIZATION ; CONDITION ; RE-INITIALIZATION ) {BLOCK} foreach VAR (LIST) {BLOCK} for VAR (LIST) {BLOCK} do {BLOCK} while (CONDITION) do {BLOCK} until (CONDITION)
排序
- 按字母顺序升序:
@array = sort @array; - 按数字升序:
@array = sort {$a <=> $b } @array;
范围操作符
(1../comments/) and next; ,其中(..) 是范围操作符,表示跳过第一行到包含comments的所有行。然后and是一个逻辑操作符,表示跳过后继续进行
逻辑操作符
and可以用&&表示,or 可以用|| 表示,not用!表示
用逻辑操作符来控制流程:比如打开文件
open (FH, $filename) or die "Cannot open file $filename: $!";
快速调用匹配值
# 例如:'123456' =~ /34/ 可以匹配成功
$&就表示匹配上的内容'34'
$`表示匹配成功前面的内容'12'
$'表示匹配成功后面的内容'56'
正则表达
要想在正则表达式中体现//,可以用/\/\/\n/ ,但总感觉有点奇怪,像这种情况,可以在前面使用m,利用其它的界定符(一般我们使用/,但并不代表只有这一种界定方式),从而避免在正斜线前使用转义符,如m!//\n!
看一个例子:
while( $features =~ /^ {5}\S.*\n(^ {21}\S.*\n)*/gm ) {};这是while循环中添加的一个正则表达式匹配:如果features满足一定的要求,就执行while语句下面的内容,那么满足什么要求呢?那就需要看表达式
不要被看似复杂的结构搞晕,直接看
/ /中间包围的那部分:其中\n表示换行符,那我们先看第一个\n前面的内容:^ {5}\S.*\n表示以5个空格开头^ {5},然后跟着一个非空白字符\S,再接任意个非换行符字符.*然后第二个
\n前面的是(^ {21}\S.*\n)*表示 以21个空格开头,然后加一个非空白字符,再接任意个非换行字符。并且注意到这部分内容被小括号包围,然后跟了星号,表示小括号中的内容可有可无。因此,
/ /内部整体上说明的是: 匹配第一行以5个空格开头,下面的行中以21个空格开头,但这部分可有可无点号加量词操作符
.{num}表示字符个数(.*?)表示非贪婪匹配或最小化匹配,会尽可能匹配短的字符串(默认*是贪婪的)关于元字符:
\ | ( ) [{ ^ $ * + ? .匹配它们需要\转义,使用|进行单一匹配,使用()用于分组 使用[]设置匹配字符集(如[A-Za-z0-9]表示任意单个字母或数字,[^0-9]匹配非数字) 使用.匹配任意一个除换行符外的字符 使用?限定量词进行最小化匹配,用在* + {}后面 使用* + {MIN,} {MIN,MAX}表示量词,其中*为0次、1次或多次,+为1次或多次,{}指定几次,可选最少和最多Perl根据小括号从左到右出现的顺序来将匹配到的字符串存储到
$1、$2等特殊变量中,例如:$alphabet = 'abcdefghijklmnopqrstuvwxyz'; $alphabet =~ /(((a)b)c)/; print "First pattern = ", $1,"\n"; print "Second pattern = ", $2,"\n"; print "Third pattern = ", $3,"\n"; #First pattern = abc #Second pattern = ab #Third pattern = a另外小括号将变量括起来还可以捕获变量,例如:
#如果不加括号,变量只会存储数组中的元素个数 @array = ('one', 'two'); $a = @array; print $a; # 结果为2 #加上括号,就变成了只有一个元素的列表,结果就是数组在列表中按行求值,并且括号中有几个元素,就能捕获到数组中几个变量 @array = ('one', 'two'); ($a,$b) = @array; print "$a $b"; #结果为 one two
模式修饰
常用的模式修饰符:用于全局匹配的/g,不分区大小写的/i,将整个字符串当成单独一行的/s(忽略换行符),将整个字符串作为含有换行符的行/m (/m可以让元字符^和$ 匹配嵌在内部换行符之前和之后的位置)
搭配元字符: ^锚定开头,$锚定结尾,.匹配除了换行符以外的任意字符
例如:要对AAC\nGTT 进行匹配
use warnings;
"AAC\nGTT" =~ /^.*$/; #使用^.*$可以匹配除了换行符外的任意字符
print $&, "\n";
# 结果报错
# 原因就在于:在中间出现了一个换行符,默认的模式修饰不能识别,因此可以转换成m修饰,会从字符串开头一直匹配到第一个换行符(即便出现在内部)
"AAC\nGTT" =~ /^.*$/m;
print $&, "\n";
#结果输出AAC,$&就是匹配到的内容
# 如果用/s,就可以匹配包括换行符在内的所有
"AAC\nGTT" =~ /^.*$/s;
print $&, "\n";
# AAC
# GTT
输出
printf:示例:
my $first = '3.14159265'; my $second = 76; my $third = "Hello world!"; printf STDOUT "A float: %6.4f An integer: %-5d and a string: %s\n", $first, $second, $third; #结果生成 A float: 3.1416 An integer: 76 and a string: Hello world!基本的指令就是
prinft % 标识 转换指令符,转换指令符就是说明输出的内容格式,包括:用于浮点数的f、整数d、字符串s。另外在%与转换指令符之间还有标识,对应的是最小字段宽度、精度、长度。例如:
%6.4f表示输出一个浮点数,最小宽度是6个字符,小数部分是四位,于是可以看到,在结果的3.1416前空了1位,就是为了满足6个字符长度的要求;%-5d指定输出整数,字段宽度为5,-表示数字居左
子程序
子程序可以重复使用,可以单独进行测试,子程序本身可以调用其他子程序,总而言之,子程序就是只做一件事并把它做好,然后我们要做的就是:组合它们来完成整个任务
编写子程序示例
子程序一般包括:sub、子程序名字、大括号代码块
子程序从@_ 数组中收集参数值,然后赋给子程序的变量(子程序的变量名可以和主程序重名,并不冲突)。如果只在子程序中写my ($dna)而不加= @_,那么子程序就会使用主程序的同名变量或者报错找不到新变量
sub addbase {
my ($dna) = @_; #perl的特殊数组是“_”。my用来声明一个新变量,只在包裹的代码块中有效
$dna .= 'ATGC';
return $dna
}
切记:子程序中定义任何变量,都要使用my ,可以在程序开始添加use strict; 提醒我们使用my定义变量
例如:计算DNA中G的数目
#!/usr/bin/perl -w
use strict; #强制声明变量
# 主程序
# 定义变量USAGE作为程序帮助文档,其中包含所有命令行参数[这里包括了$0和DNA]
# $0也是特殊变量,表示这个perl脚本名;DNA指的是需要的DNA序列
my ($USAGE) = "USAGE:$0 DNA\n\n"
# 数组变量@ARGV是内置变量,其中包含输入的参数;如果没有输入参数就打印程序帮助文档USAGE
unless (@ARGV){
print $USAGE;
exit;
}
my ($dna) =$ARGV[0]; #提取数组第一个元素,注意要换成$提取(perl的坐标从0开始)
my ($sum_Gs) = countG($dna);
print "\nThe DNA $dna has $sum_Gs G\'s in it!\n\n";
exit;
#子程序
sub countG{
my ($dna) = @_;
my ($count) = 0;
$count = ($dna =~ tr/Gg//);
return $count;
}
传递数据给子程序
值传递:(参数比较简单)主程序参数复制后传给子程序,子程序中这部分值改变并不会影响主程序相应参数的值,例如:
#!/usr/bin/perl -w use strict; my $i=2; subvalue($i); print "In the main program, the result is $i\n"; exit; sub subvalue{ my ($i)=@_; $i += 100; print "In the sub program, the result is $i\n"; } #In the sub program, the result is 102 #In the main program, the result is 2引用传递:(参数比较复杂)可以向子程序中传递标量、数组等各种形式的参数,子程序中所做的任何事都改变主程序中的对应参数。重点是:在引用的变量名前加一个反斜线
#!/usr/bin/perl -w use strict; #主程序 my @i = ('1','2','3'); my @j = ('a','b','c'); print "Mian-1 program: i = "."@i\n"; print "Mian-1 program: j = "."@j\n"; subvalue(\@i,\@j); print "Main-2 program: i = "."@i\n"; print "Main-2 program: j = "."@j\n"; exit; #子程序 sub subvalue{ my($i,$j)=@_; print "Sub program: i = "."@i\n"; print "Sub program: j = "."@j\n"; push(@$i,'4'); shift(@$j); } #Mian-1 program: i = 1 2 3 #Mian-1 program: j = a b c #Sub program: i = 1 2 3 #Sub program: j = a b c #Main-2 program: i = 1 2 3 4 #Main-2 program: j = b c关于指针:只有一个
$的变量都是标量变量的指针,作用仅仅是当一个中间桥梁的作用;如果要直接表示指向的那个标量变量,需要用两个$,同理,如果要表示数组变量就用@$,表示散列变量%$
重复使用的子程序—模块/库
子程序确实很方便,但是一些比较好的子程序需要应用到许多脚本中,一旦需要修改就需要修改所有脚本中的子程序,这是非常麻烦的。因此,如果一开始将子程序放到一个方便使用的文件中,如果改动就只改一次,然后可以应用到所有的脚本就好了。因此,模块(库)就诞生了。
例如,我们有一个库叫beginperl.pm(后缀名是perl module的意思),里面存放了所有的子程序。【需要注意的是,.pm文件的最后一行要是1;】。
使用库的时候,直接使用use beginperl; 【注意:这条语句放在靠近use strict;的地方;库的名字不带后缀;库如果和脚本不在一个目录下,需要绝对路径】
例如:File::Find模块帮助定位文件和目录,找到一个好的模块可以减少编程时间
应用
一:提取Genbank序列和注释信息
先对Genbank的信息分布有个大体了解,比如序列信息在最后,从ORIGIN开始到最后一行的//,注释信息是ORIGIN之前的信息
**基本思路:**加载文件=》遍历数组(文件每一行)=》先匹配// 得到最后一行行号=》再匹配ORIGIN得到分界线行号【之所以先匹配//是因为如果先匹配分界线的话,一旦匹配上就不会再继续进行了,也就得不到最后一行的行号了】=》序列就是ORIGIN行向下,注释就是ORIGIN行向上
#前提得到Genbank文件@Genbankfile
#sub open_file
# given the filename, return the filehandle
sub open_file {
my ($filename) = @_;
my $fh;
unless (open($fh, $filename)){
print "cannot open\n";
exit;
}
return $fh;
}
# 这里只是列一个基本思路
my $linenumber = 0;
my $end;
my $origin;
foreach my $line (@Genbankfile){
if ($line =~ /^//\n/) {
$end = $linenumber;
last;
}elsif ($line =~ /^ORIGIN/){
$origin = $linenumber;
}
$linenumber++;
}
@annotation = @Genbankfile[0..($origin-1)];
@seq=@Genbankfile[($origin+1)..($end-1)];
关于读取文件:可以在命令行给出要打开的文件名(例如
perl a.txt test.pl),并在perl脚本中第一行使用<>就可以表示读取文件,例如while (<>) {操作}
二、Blast
非编程相关内容
不论是网页还是本地运行的blast,都是基于字符串匹配程序,利用字符串匹配来查找相似,来作为同源的一个指标。相似度(similarity)可以用percent identity来衡量,就是说查询的序列和数据库中的某个序列对应区域完全匹配的碱基数目,它会找到等价密码子或者不改变蛋白功能并具有相似属性的氨基酸残基之间的匹配,表示一个保守的程度。说到序列同源(homology) ,它是指序列在进化上相关,要么同源,要么不同源,没有同源度这种说法
Blast的运行主要基于:原始值S、打分算法、数据库属性。原始值(Raw score S)是对相似性和匹配大小的度量。另外在Blast的结果中还能看到:按照E值排序的hits数。一个匹配的E值/期望值(E value/Expect value)表示一个随机生成的具有同样大小和数据库构成的字符串匹配几率(其中允许空位存在)。E越接近0,表示越不可能随机发生。因此,E越小,匹配结果越好 。一般E小于1是一个比较可靠的结果
资料:
http://barc.wi.mit.edu/education/bioinfo2003/perl_bioinfo.html
https://github.com/Yixf-Education/course_Perl/tree/master/book