005-R-GenomicRanges使用
刘小泽写于18.6.22
本文包含了Rstudio安装、bioconductor使用以及常用R包GenomicRanges的介绍
R & RStudio安装:
官网下载 https://www.rstudio.com/products/rstudio/download/#download If you don’t already have R, download it here
Bioconductor使用:
官网链接https://www.bioconductor.org/
官网主页:
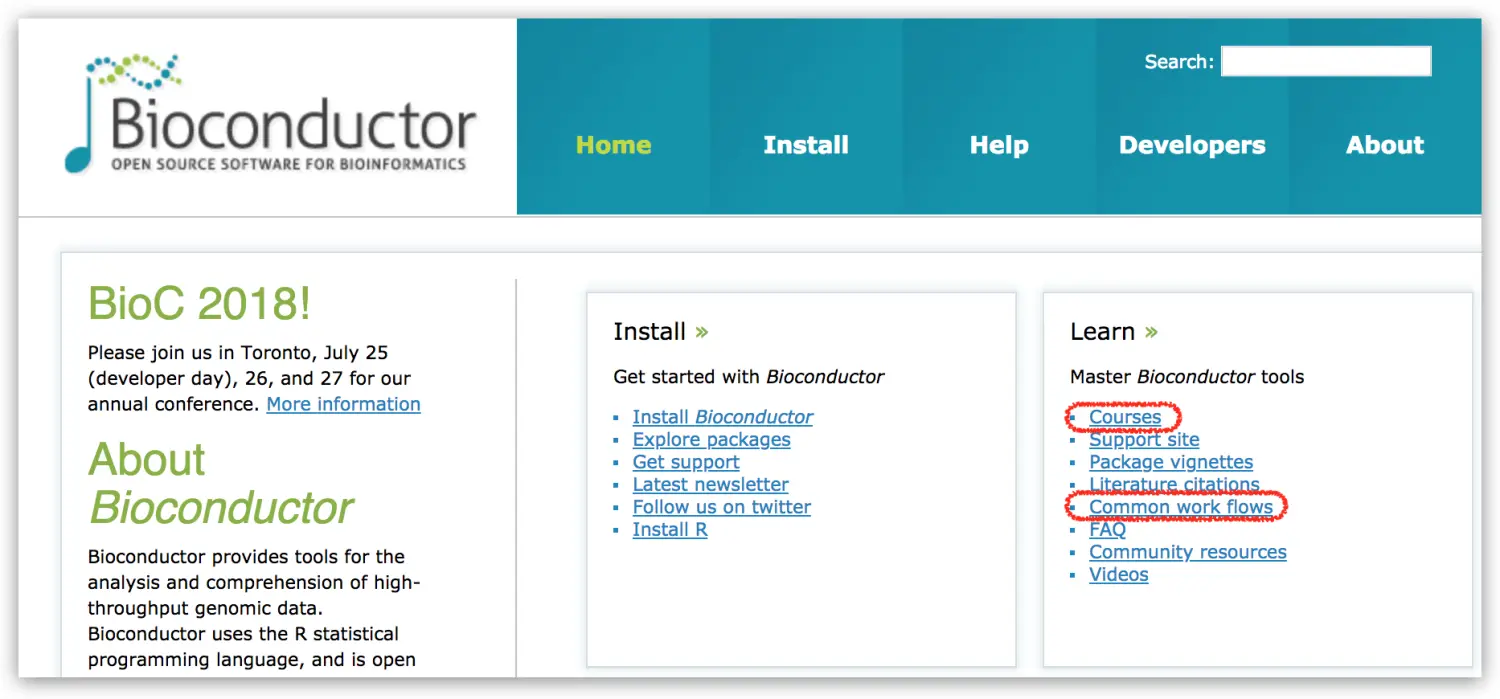
主要关注Courses、Common work flows这两项内容
Courses列出了各种Talk、Conference的slides,方便学习
最干的干货当属Common work flows,跟着教程一步步走,你也能做出最后的图片。并且涵盖的内容很广,基因表达、注释、蛋白质组学、单细胞测序、基因组变异检测等等,全部都有规范详尽的pdf指导
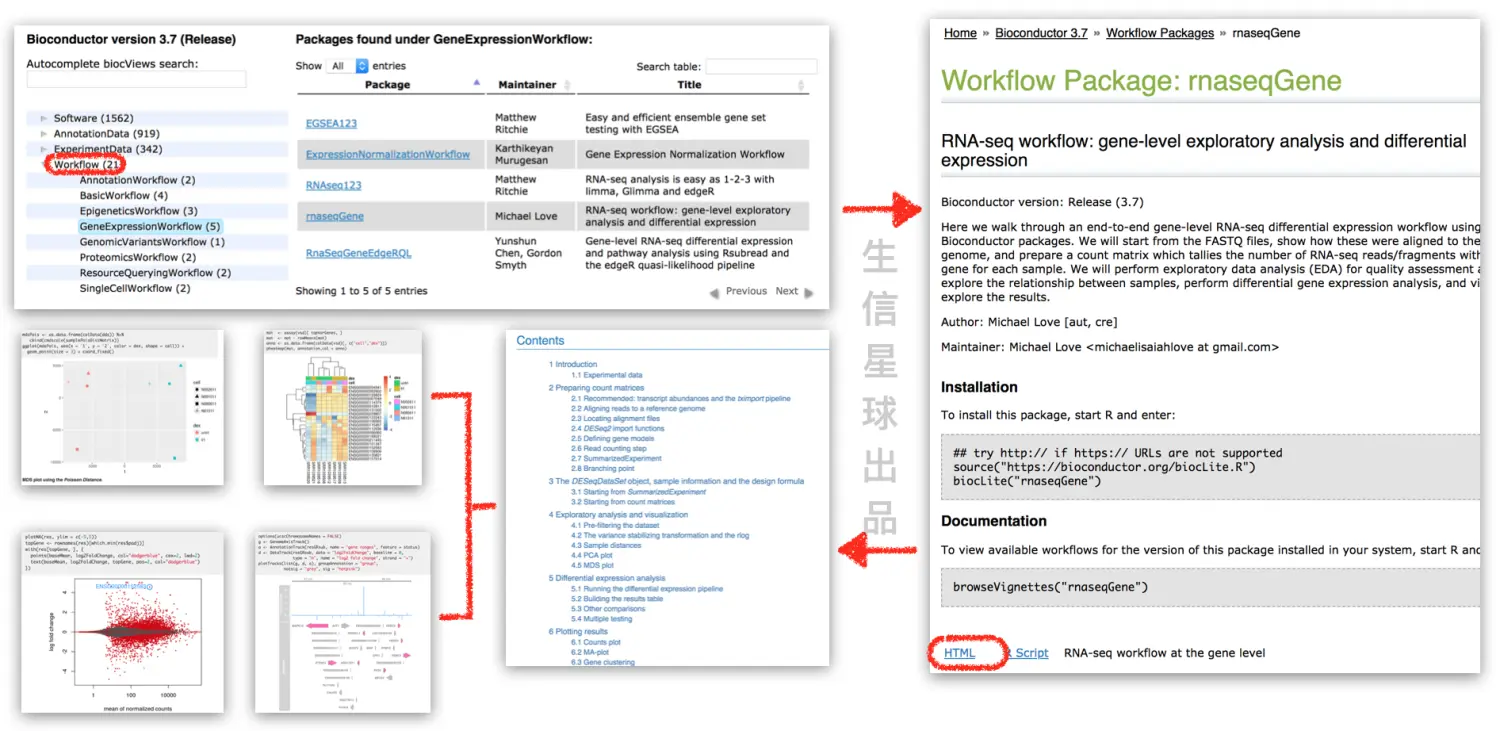
Bioconductor的核心主要包括以下几个包:
- GenomicRanges: 表示基因组范围
- GenomicFeatures: 表示基因模型和基因组其他特性(genes, exons,UTRs,transcripts等)
- Biostrings和BSgenomes:对基因组序列进行操作(比如有些包可以从ranges中提取序列)
- rtrcklayer: 读取常见的生物信息数据格式,BED,GTF/GFF,和WIG
什么是GenomicRanges?
官网给出的解释
This package defines general purpose containers for storing and manipulating genomic intervals and variables defined along a genome.
This package lays a foundation for genomic analysis by introducing three classes (GRanges, GPos, and GRangesList), which are used to represent genomic ranges, genomic positions, and groups of genomic ranges.
这个R包可以用来对基因组位置进行操作【基因组位置(genomic range/intervals)由染色体号、起始和结束位点、链方向组成,每个基因组版本都有特定的位置信息】。 它可以定义序列名称,包括起始点及终止点的长度信息,正负链,或者他们的score值和GC值等。随着NGS的大规模应用,原来主打microarray的bioconductor也开始向NGS转移。GR包和IRanges、Biostrings一起构成了NGS数据的基础。
下载安装:官网主页右上角search中输入GenomicRanges就能出来这个包的介绍
使用Rstudio安装
source(“https://bioconductor.org/biocLite.R") biocLite(“GenomicRanges”)
加载library(“GenomicRanges”)
1. GRanges:存储基因组features位点
> gr <- GRanges(seq = Rle(c("chr1", "chr2","chr3","chr1"), c(2,3,3,2)), + ranges = IRanges(101:110, end = 111:120, names = head(letters,10)), # 这里创建ranges也可以指定起始位点start和宽度width,就不需要end了 + strand = Rle(strand(c("-", "+", "*","-","+")), c(2,3,1,2,2)), + score = 1:10, + GC = seq(0,1,length=10))创建了10条序列的信息:
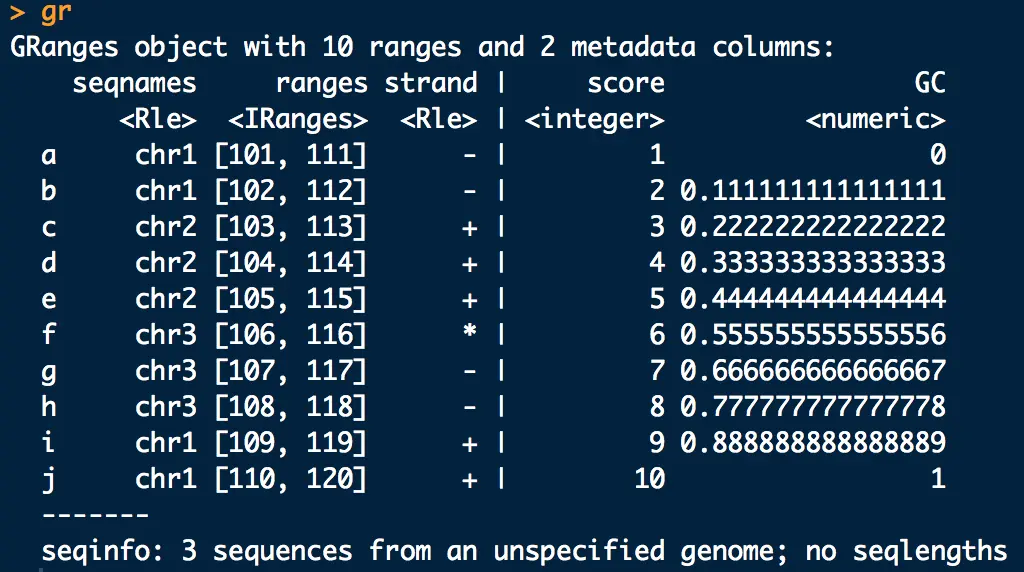
以
|为界分为左右两块区域: 左边区域:【必选】基因组坐标信息(seqnames, ranges, strand);用granges(gr)查看 右边区域:【可选】注释信息(score, GC 等); 用mcols(gr)查看,用mcols(gr)$score查看具体项width()统计基因组序列长度分布;length()计算行数;names()查看最前列的名称1.1 gr拆分与重组
拆分:
sp <- split(gr, rep(1:2,each=5))意思是分成两组,一组五个重复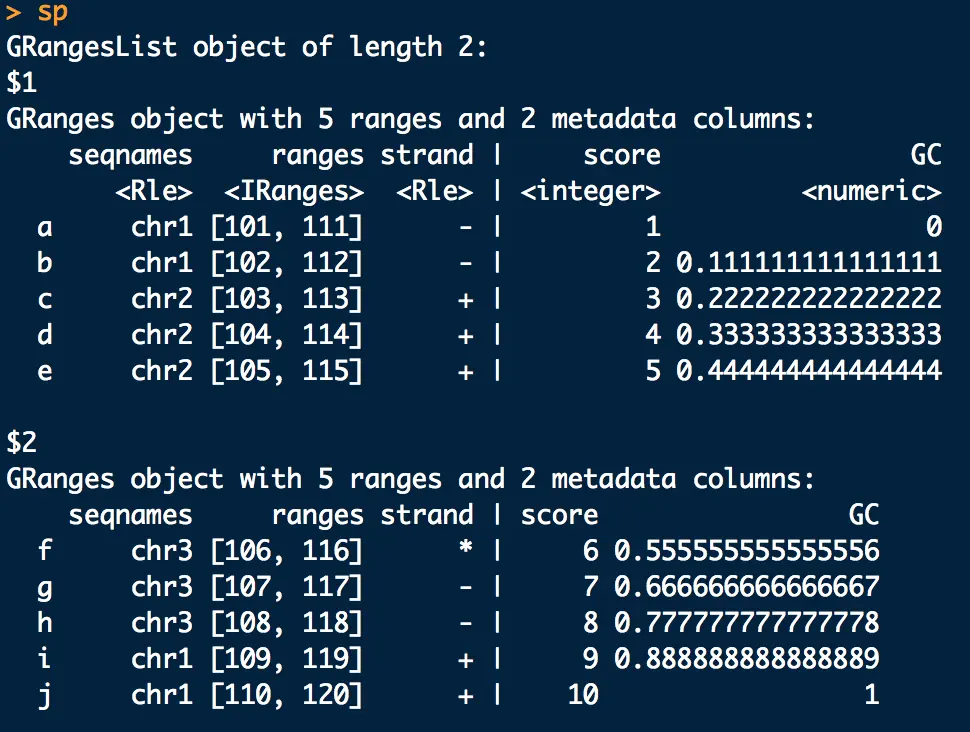
重组:
c(sp[[1]],sp[[2]]), 当然c()也可以连接两个GRange object 重组后去重:unique()1.2 取子集
1.2.1
gr[2:3]得到2、3列;gr[2:3, "GC"]多含了GC这一项1.2.2 按names划分成单行
singles <- split(gr, names(gr),对单行操作:
#复制 rep(singles[[2]], times = 3) #倒置 rev(gr) #选择特定区域head(gr, n=2) 得到前两行,tail 从后向前数 #选择特定行 window(gr, start=2,end=4),选择了2-4行 #选择跨区域特定行gr[IRanges(start = c(2,7), end = c(3,9))] ,选择2-3行 & 7-9行1.3 序列间隔操作【对于一个gr.obj而言】
1.3.1 基本操作:start、end、width(=end-start)、range(将一条链上相连的区域组合在一起进行统计)
1.3.2 flank:翻译为侧翼,可用来找上下游特定区域**(详见3 .3)**
1.3.3 shift: range整体平移**(详见3.4)** ; resize: 改变range宽度【+链修改end位置;-链修改start位置】
1.3.4 reduce:比对ranges,然后merge overlap (详见3.4下方图)
1.3.5 gaps: 寻找range之间的gap
1.3.6 disjoin(): 将range变为non-overlapping (详见3.6)
1.3.7 coverage:统计所有ranges中overlap的数量
1.4 比较多个gr.obj
1.4.1 union(gr1, gr2) 寻找并集;insersect()寻找交集;setdiff()寻找补集
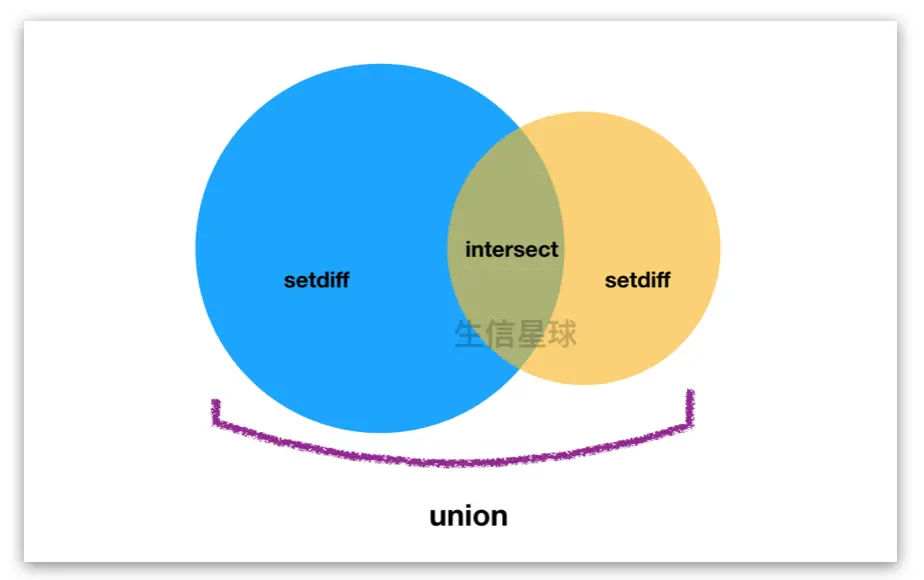
1.4.2 当两个gr.obj的行数相同时,即他们是parallel的关系,使用punion() / pintersect() / psetdiff()
2. GRangesList
一些比较重要的基因组特征信息,比如转录本它构成了外显子,但是这些区域是分散的。单看一个是不够的,聚合在一起表达时才能揭示一些信息。如何将这些分散的特征信息聚集在一起?
GRangeList就能完成这个工作:
2.1 比如现在有两个转录本,我们需要先把它们组合在一起
gr1 <- GRanges( seqnames = "chr2", ranges = IRanges(103,106), strand = "+", score = 5L, GC = 0.45) gr2 <- GRanges( seqnames = c("chr1", "chr1"), ranges = IRanges(c(107,113), width = 3), strand = c("+", "-"), score = 3:4, GC = c(0.3,0.5) ) grl = GRangesList("txA" = gr1, "txB" = gr2)就得到这样的list:

2.2 查看基本信息
seqnames(grl)、ranges(grl)、strand(grl)length()、names()输出list的信息;seqlength()输出list子集的长度信息elementNROWS(grl)输出各子集的行数,相对于data_frame中的lapply要更快isEmpty(grl)检查list中是否包含空objectmcols(unlist(grl))查看list下各object的metadata(如score/ GC等)的信息2.3 合并list
先合并
grl3 <- c(grl1, grl2), 再按gr3中各object的名称重组regroup(grl3, names(grl3))2.4 取子集
[]返回的是list其中一个object; [[]]返回的是object中的某行
对子集进行操作:
head(grl, n=1)、tail(同head)、rev(grl)、rep(grl[[1]], times =3) window(grl, start =1, end=1)start和end代表list中object的顺序,这里只取出了第一个object2.5 list集合中的多重操作
2.5.1
sapply(grl, length)对list中各个obj求长度【lapply也可以,只是输出格式为列表】 【sapply的 ‘s’ 意思是simple; lapply的 ‘ l’ 意思是list】2.5.2
mapply(c, grl, grl2)对多个list中的obj分别进行合并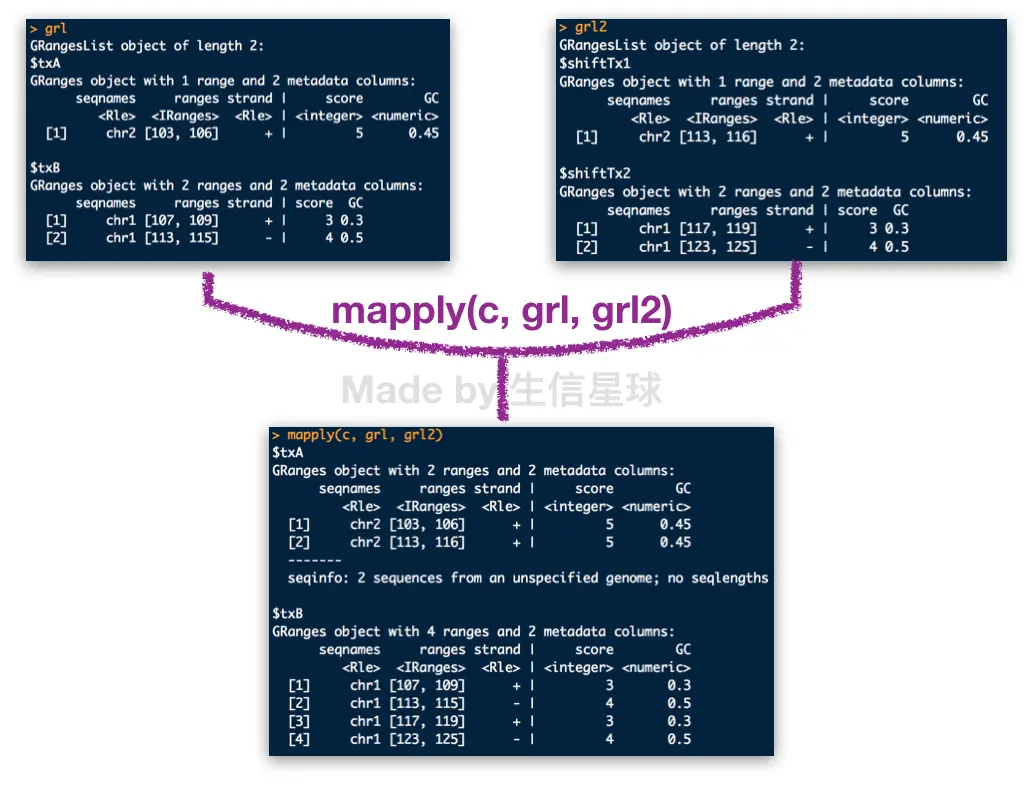
2.5.3 去list中冗余
Reduce(c, grl),会把所有的obj们合并在一起2.5.4 想对list中某一个object进行操作
流程是:unlist -> command -> list# 例如,想在object中增加一列meta信息log_socre,求socre的对数 gr <- unlist(grl) gr$log_score <- log(gr$score) grl <- relist(gr, grl)

3. 比较实用的部分
相对于data_frame,gr是运行的更快的
3.1数据过滤:
3.1.1 比如想得到score小于5的,
gr[gr$score<5]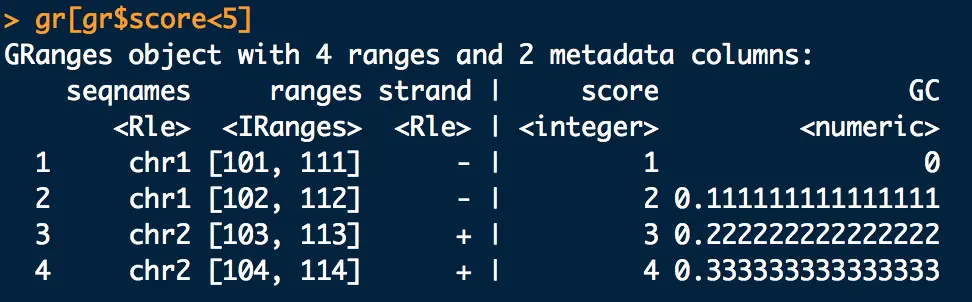
3.1.2 在此基础上也可以增加过滤条件,比如得到score大于5并且GC含量小于0.8的
gr[gr$score>5 & gr$GC<0.8]3.1.3 取出strand为正链的
gr[strand(gr) == "+"]; 除了正链其余都要取就用gr[strand(gr) != "+"]3.1.4 假如数据是全基因组,想分析2号染色体数据 gr[seqnames(gr) == ‘chr2’]
3.2 数据排序
sort()按基因组seqname顺序,先排正链+,再排负链-,最后排*(正链或负链)order()根据某项可选内容(|右侧)排序,gr[order(gr$GC,decreasing = T)]这是根据GC含量降序排序3.3 flank
问题:假如gr2中的每一行是一个基因,如果想寻找基因上游10bp的region,怎么做?
【+链起始位置向前数,-链终止位置向后数】
先看一下gr2
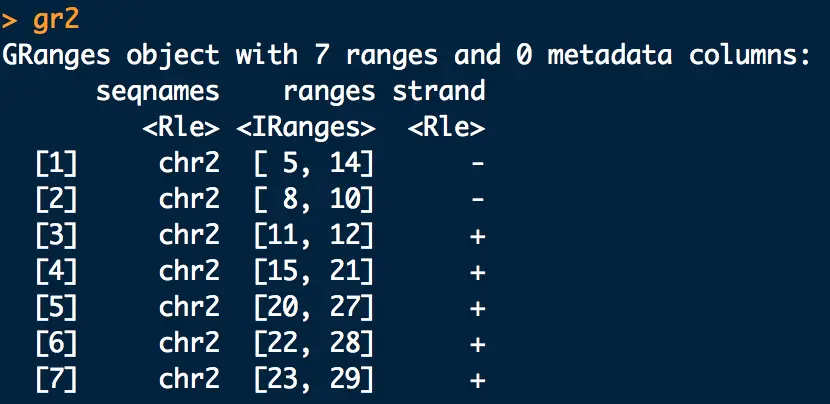
flank(gr2,width = 10)得到的就是上游region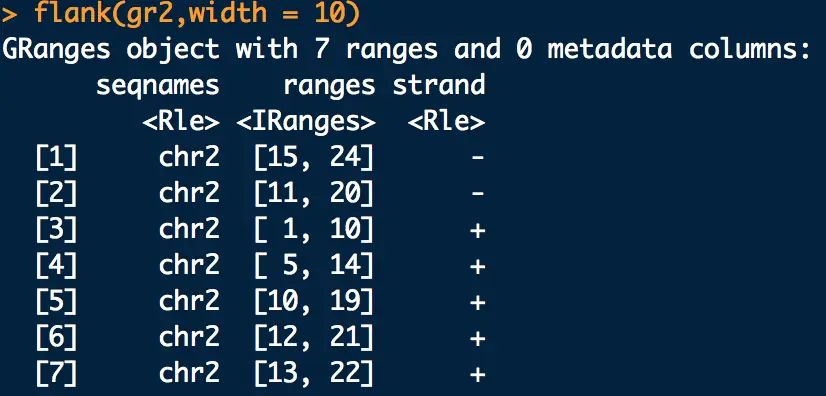
那么取下游怎么取?
gr3 <- flank(gr2,width = 10, start = F)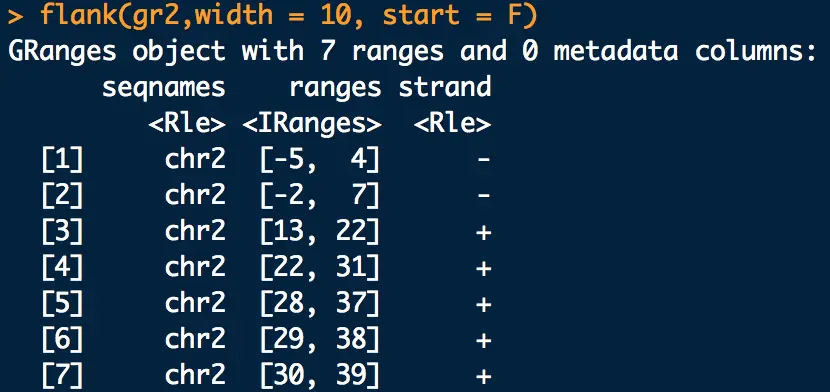
起始位置出现了负数,只需替换成1即可, 这就是下游region。
start(gr3[start(gr3) < 1]) = 1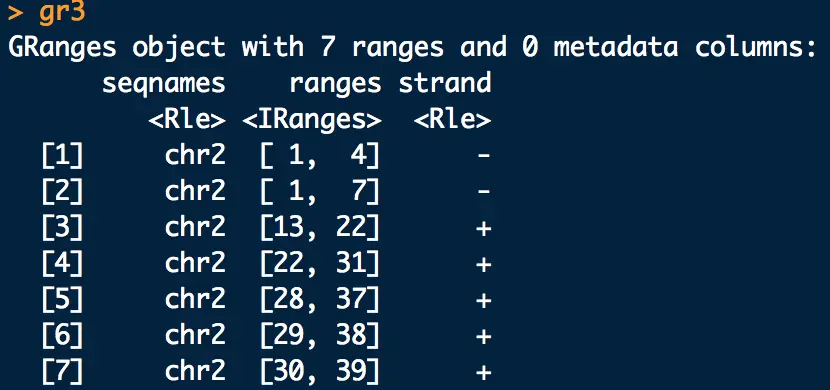
做这个工作的意义何在?
分析上游upstream其实就是等同于找promoter region 【启动子存在于上游2k – 下游100/200bp,并非一定在TSS (转录开始的位置)之前才是】; 分析下游downstram 可以寻找UTR 【UTR长度不固定,3‘-UTR/ trailer存在alternative poly A 调控元件。UTR越长,一般能target的microRNA也越长。】
再简短介绍下miRNA:microRNA(miRNA)是一种长度约为22核苷酸(nt)的非编码RNA,其主要通过碱基互补与靶mRNA的3’端非翻译区(3’UTR)结合,导致靶mRNA降解或抑制蛋白质的合成,在转录后水平调节基因的表达

流程是这样的:
gene--> gr_oblect --> flank(gr_object, width = 2000)就能找出启动子区域当然,GRange自带了
promoter()可以完成flank找启动子的工作,并且直接在上、下游都同时寻找promoters(gr2, upstream = 2000, downstream = 10)3.4 shift 无论正链还是负链都进行整体平移
- 正向平移
shift(gr2,shift = 10) - 负向(向染色体上游)平移
shift(gr2,shift = -10)
介绍下面前先看一张图,介绍了shift、reduce、disjoin的关系 可以看出,shift的平移, reduce合并overlap, disjoin拆分overlap
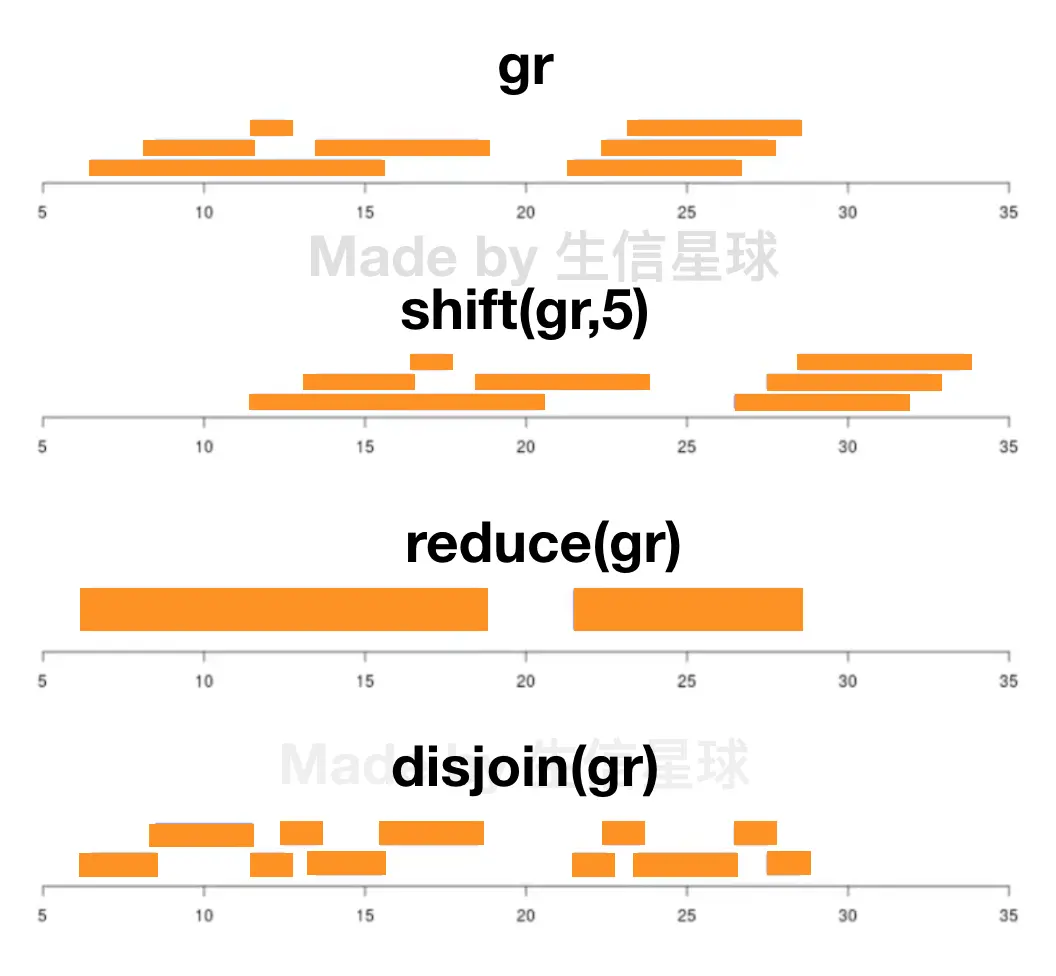
3.5 reduce
作用:假设有一个gff/gtf文件,gtf/gff -> GRange -> reduce() -> total(exon)
3.6 disjoin
作用: gtf/gff -> GRange -> disjoin -> remove overlap region -> specifc region -> Alternative splicing
3.7 findOverlaps
作用:很明显,找两个region的overlap,也就是找区域交集 意义:寻找功能区间的交集是基因组学的必备内容
findOverlaps(gr1,gr2)这种是两两比较,结果也是相互比对的,还可以在括号中加上select=“first/last/arbitrary”意思是返回第一个/最后一个/随机的交集位置。或者
gr1[gr1 %over% gr2]这样找到的就是存在于gr1中的gr1与gr2的overlapex. Chip-seq中会寻找信号在promoter的富集:
1. 找到promoter区域; 2. 找到chip-seq的region; 3. 比对他们之间有没有overlap 4. 将3中的overlap同基因组的overlap比对看有没有显著富集