049-一套limma、edgeR的实战
刘小泽写于18.10.26 今晚10.15开始写
第一部分 背景介绍
选用GEO数据:GSE63310
从雌性小鼠乳腺中提取了三种类型的细胞:basal、luminal progenitor(LP-乳腺癌早期前体细胞)、mature luminal (ML),各三个重复。RNA利用Hiseq 2000+100bp单端测序;参考基因组选择mm10,Rsubread比对+featureCounts定量
转录组表达数据地址:http://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file
第二部分 数据准备
读取表达量数据 =>得到DEGList x
cd ~/Download
wget -c http://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file
tar xvf GSE63310_RAW.tar
unzip *
### 设置好rstudio 的project
files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt",
"GSM1545538_purep53.txt","GSM1545539_JMS8-2.txt",
"GSM1545540_JMS8-3.txt","GSM1545541_JMS8-4.txt",
"GSM1545542_JMS8-5.txt","GSM1545544_JMS9-P7c.txt",
"GSM1545545_JMS9-P8c.txt")
read.delim(files[1], nrow=5)
## EntrezID GeneLength Count
##1 497097 3634 1
##2 100503874 3259 0
##3 100038431 1634 0
##4 19888 9747 0
##5 20671 3130 1
#接下来利用edgeR的readDEG将所有表达矩阵读入并且组合成一个大的矩阵
library(limma)
library(edgeR)
x <- readDGE(files, columns=c(1,3)) # 只要它的Entrez ID 和count值
dim(x) # x中有表达量和样本信息,样本信息又包括了group、libsize、lane等信息
#[1] 27179 9
# 如果一开始所有的表达量都存在一个矩阵中,那么直接用DEGList()转换就好
整理样本分组信息 =》 对DEGList x的列进行操作
下游分析之前,需要将整合的矩阵的列名与实验样本设计关联起来(包括不同的分组信息以及不同组的重复信息)
比如:细胞的basal、LP、ML分组;基因型的wild-type、knock-out分组;表型的disease、status、sex、age分组、样本处理的drug、control分组、关于批次Batch的不同样本采集、测序日期、测序lane等
这里的DEGList中包含的样本信息有:细胞类型、批次信息(测序的lane),每种信息都是因子型factor变量,但是各自因子的level不同
# 先简化GEO的ID名称,也就是把GSM去掉
samplenames <- substring(colnames(x), 12, nchar(colnames(x)))
samplenames
##[1] "10_6_5_11" "9_6_5_11" "purep53"
##[4] "JMS8-2" "JMS8-3" "JMS8-4"
##[7] "JMS8-5" "JMS9-P7c" "JMS9-P8c"
colnames(x) <- samplenames
#然后我们可以自己设计分组信息
group <- as.factor(c("LP", "ML", "Basal", "Basal", "ML", "LP",
"Basal", "ML", "LP"))
x$samples$group <- group
#自己添加lane 信息
lane <- as.factor(rep(c("L004","L006","L008"), c(3,4,2)))
x$samples$lane <- lane
x$samples
## files group lib.size norm.factors lane
##10_6_5_11 GSM1545535_10_6_5_11.txt LP 32863052 1 L004
##9_6_5_11 GSM1545536_9_6_5_11.txt ML 35335491 1 L004
##purep53 GSM1545538_purep53.txt Basal 57160817 1 L004
##JMS8-2 GSM1545539_JMS8-2.txt Basal 51368625 1 L006
##JMS8-3 GSM1545540_JMS8-3.txt ML 75795034 1 L006
##JMS8-4 GSM1545541_JMS8-4.txt LP 60517657 1 L006
##JMS8-5 GSM1545542_JMS8-5.txt Basal 55086324 1 L006
##JMS9-P7c GSM1545544_JMS9-P7c.txt ML 21311068 1 L008
##JMS9-P8c GSM1545545_JMS9-P8c.txt LP 19958838 1 L008
整理基因注释信息=》 对DEGList x的行进行操作
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite("Mus.musculus")
library(Mus.musculus)
geneid <- rownames(x)
genes <- select(Mus.musculus, keys=geneid, columns=c("SYMBOL", "TXCHROM"),
keytype="ENTREZID") #转换基因id,用clusterProfiler的bitr函数也可以;另外这个还增加了染色体信息
head(genes)
## ENTREZID SYMBOL TXCHROM
##1 497097 Xkr4 chr1
##2 100503874 Gm19938 <NA>
##3 100038431 Gm10568 <NA>
##4 19888 Rp1 chr1
##5 20671 Sox17 chr1
##6 27395 Mrpl15 chr1
需要注意的是:Entrez ID可能并不是和基因信息一一匹配的,可能同样的ID会匹配到不同染色体,因此需要检查有没有重复出现的Entrez ID,以保证注释和我们的DEGList之间的基因顺序是一致的
dup <- genes$ENTREZID[duplicated(genes$ENTREZID)]
genes[genes$ENTREZID %in% dup,][1:5,]
# 果然发现了不同染色体上重复的ID
## ENTREZID SYMBOL TXCHROM
##5360 100316809 Mir1906-1 chr12
##5361 100316809 Mir1906-1 chrX
##9563 12228 Btg3 chr16
##9564 12228 Btg3 chr17
##11350 433182 Eno1b chr4
# 然后把重复的基因挑出来【重复的只统计一次】
mat <- match(geneid, genes$ENTREZID)
genes <- genes[mat,]
genes[genes$ENTREZID %in% dup,][1:5,]
这时在看x这个DEGList,就是一个标准的包含原始count数据、样本信息的表达量矩阵
第三部分 数据预处理
原始数据的转换
进行差异表达一般都不会用raw counts的,因为存在测序深度、文库大小的差别,这样的结果是不准确的
一般的做法是:利用标准化算法,如CPM(counts per million), log-CPM (log2-counts per million), RPKM (reads per kilobase of transcript per million), FPKM(fragments per kilobase of transcript per million)等去除文库大小、深度的影响。和RPKM、FPKM不同的是,CPM和log-CPM不需要考虑feature length的差异,也就是说基因长度在统计时被当成常数,只考虑不同处理下的不同,而不会受长度的影响
cpm使用cpm()函数;RPKM使用rpkm函数,都属于edegR
cpm <- cpm(x)
lcpm <- cpm(x, log=TRUE)
去掉不感兴趣的基因
所有的数据集中都会存在表达的和不表达的基因,我们感兴趣的是在一个条件下表达,另一个条件不表达的。
看下有多少基因在所有样本中表达量都为0
table(rowSums(x$counts==0)==9)
##
## FALSE TRUE
## 22026 5153
表达量都为0的占比达到了19%
过滤基因=>标准就是cpm至少一组或整个实验中有三个样本大于1
keep.exprs <- rowSums(cpm>1)>=3
x <- x[keep.exprs,, keep.lib.sizes=FALSE]
dim(x)
作图检查:虚线就是cpm为1(即log cpm为0的时候)作为判断阈值,看超过这个线的有几个
library(RColorBrewer)
nsamples <- ncol(x)
col <- brewer.pal(nsamples, "Paired")
par(mfrow=c(1,2))
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.21), las=2,
main="", xlab="")
title(main="A. Raw data", xlab="Log-cpm")
abline(v=0, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
lcpm <- cpm(x, log=TRUE)
plot(density(lcpm[,1]), col=col[1], lwd=2, ylim=c(0,0.21), las=2,
main="", xlab="")
title(main="B. Filtered data", xlab="Log-cpm")
abline(v=0, lty=3)
for (i in 2:nsamples){
den <- density(lcpm[,i])
lines(den$x, den$y, col=col[i], lwd=2)
}
legend("topright", samplenames, text.col=col, bty="n")
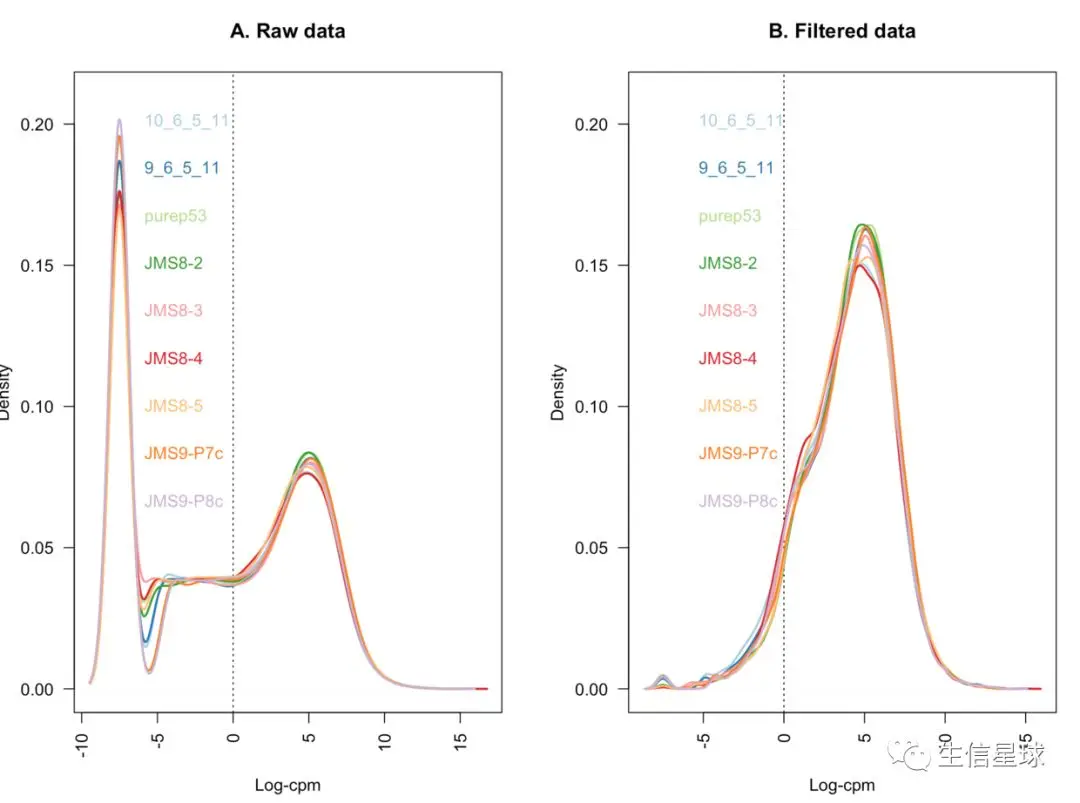
基因表达分布标准化
在准备试验样品或者测序的过程中,外界因素非常可能会引入误差,影响样本的基因表达水平。比如:第一批测序的样本可能比第二批测序的深度要深。标准化就是为了让每个样本的表达量分布在整个实验中是相似的
如何判断是否标准了呢?
可以通过密度图density或者箱线图boxplot,比如上图就是密度分布,其中B图的log-CPM分布就比较一致,并且都在阈值的右侧
如何标准化?
使用TMM(trimmed mean of M-values)算法,利用edgeR中函数calNormFactors()
标准化用到的normalisation factors 就在DEGList中,x$samples$norm.factors.调取
x <- calcNormFactors(x, method = "TMM")
x$samples$norm.factors
## [1] 0.896 1.035 1.044 1.041 1.032 0.922 0.984 1.083 0.979
画一个箱线图可以看到标准化前后差异【模拟数据】
#模拟一个数据
x2 <- x
x2$samples$norm.factors <- 1
x2$counts[,1] <- ceiling(x2$counts[,1]*0.05) #第一个样本count缩小到原来5%
x2$counts[,2] <- x2$counts[,2]*5 # 第二个样本扩大到原来5倍
#画图
par(mfrow=c(1,2))
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="A. Example: Unnormalised data",ylab="Log-cpm")
x2 <- calcNormFactors(x2)
x2$samples$norm.factors
## [1] 0.0547 6.1306 1.2293 1.1705 1.2149 1.0562 1.1459 1.2613 1.1170
lcpm <- cpm(x2, log=TRUE)
boxplot(lcpm, las=2, col=col, main="")
title(main="B. Example: Normalised data",ylab="Log-cpm")
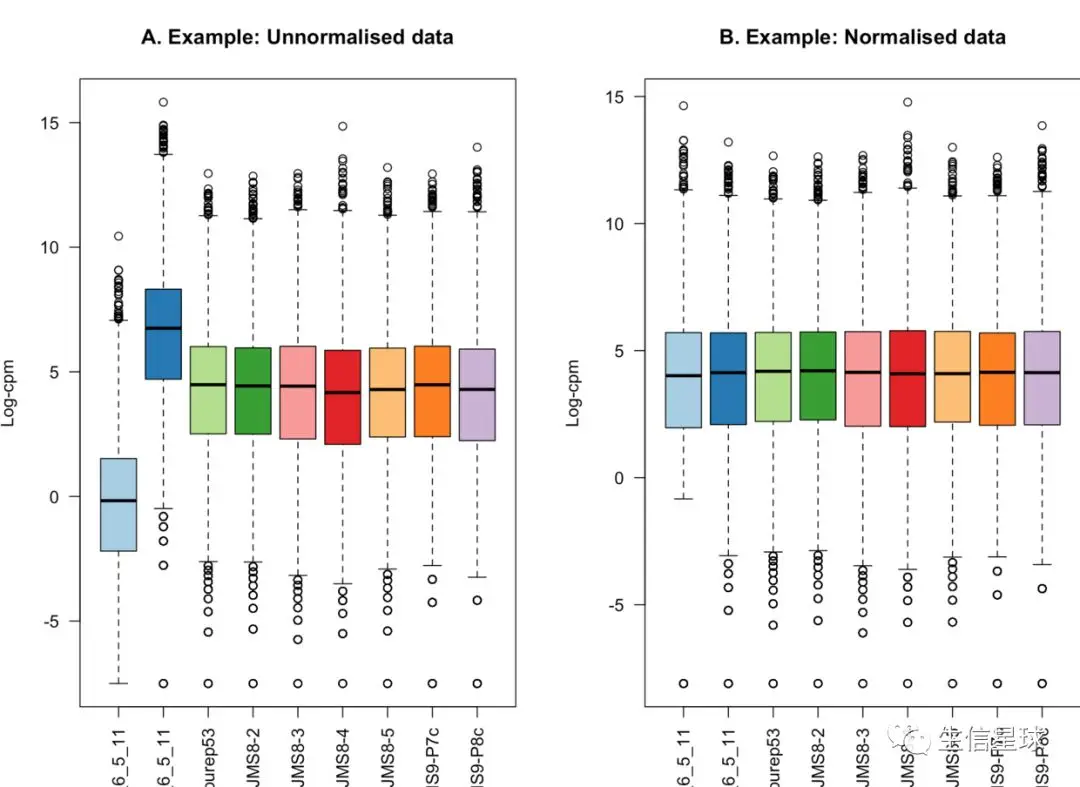
来自10.28 这些天一直在写论文
非监督式样本聚类
利用MDS(multidimensional scaling 多维坐标)作图,将样本的异同点展示出来,因此在正式差异分析之前,可以大体给我们一个判断,哪些差异表达可以被检测到。理论上,同样背景的样本应该聚在一起(比如技术重复),如果其中有的脱离群体,那么就可能是个错误,有时就需要把那个偏离样本去掉。limma包的plotMDS 函数就是干这个的,画出的第一个维度往往解释了最大的差异来源。
lcpm <- cpm(x, log=TRUE)
par(mfrow=c(1,2))
# 样本
col.group <- group
levels(col.group) <- brewer.pal(nlevels(col.group), "Set1")
col.group <- as.character(col.group)
col.lane <- lane
levels(col.lane) <- brewer.pal(nlevels(col.lane), "Set2")
# lane
col.lane <- as.character(col.lane)
plotMDS(lcpm, labels=group, col=col.group)
title(main="A. Sample groups")
plotMDS(lcpm, labels=lane, col=col.lane, dim=c(3,4))
title(main="B. Sequencing lanes")