048-RNA-seq数据的基因共表达网络分析
刘小泽写于18.10.24 最近开始写第一篇英文小论文,练练手,为将来写英文大论文做准备
目录
1 背景 Background
- Types of biological networks
- Motivation for using co-expression networks
- Network inference and reverse engineering
- Basic graph terminology and data structures
- Steps for building a co-expression network
- Optimizing parameters for network construction
2 共表达流程 Tutorial
- Preparing RNA-seq data for network construction
- Building a co-expression network
- Detecting co-expression modules
- Annotating a co-expression network
- Visualizing network
正文开始
1.1 Types of biological networks
生物网络可以包含不同的数据类型,用点(node)和边(edge)区分。常见的网络类型:
- 蛋白互作(PPI) 表示蛋白之间物理联系,它们几乎占据了细胞生物过程的中心位置。蛋白作为点,用无向的线连接
- 代谢网络 主要表示生化反应,有助于生物生长、繁殖、维持结构。点是代谢产物,并用有向的箭头表示代谢过程或特定反应的调节作用
- 基因互作 不同的点表示不同基因,描述它们功能相关性;可以根据基因的背景知识来推断线的方向
- 基因/转录调控 表示基因表达是如何被调控的;点是基因或转录因子,它们之间的关系也是定向,例如Reactome、KEGG等数据库中表示基因调节的关系
- 细胞信号 点表示通路中的物质,如蛋白、核酸或其他代谢物

1.2 Motivation for using co-expression networks
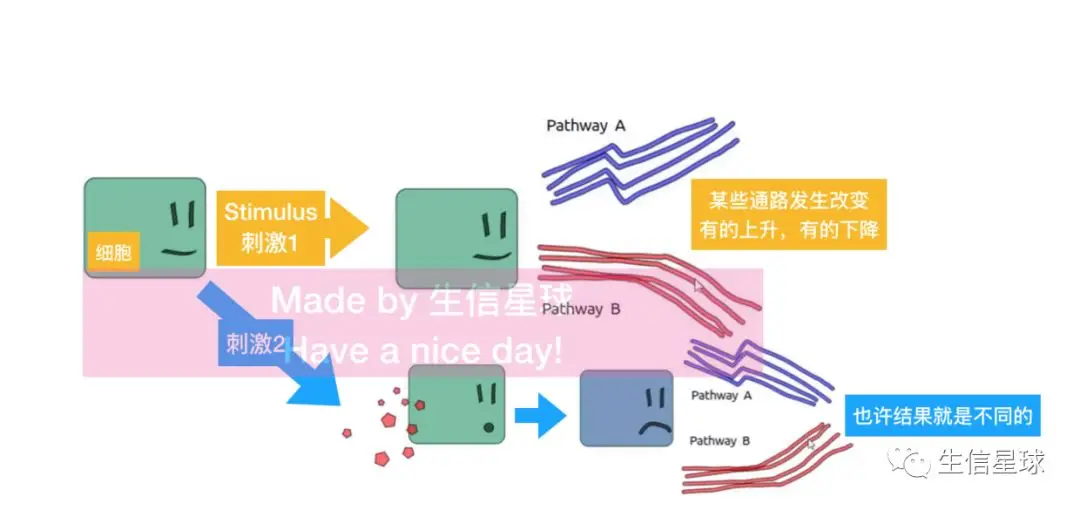
**看图说话:**某个细胞受到刺激1,也许它的A通路就会上调表达,B通路下调,结果可能比刺激前还要理想;
受到另一种刺激2后,A通路下调,B通路上调,那么可能就比较糟糕
通过共表达网络,就可以探索A、B通路是如何被调控的,以及背后基因的相互关系;另外,互作的基因一般都参与同样的生物途径
一般来讲,探索基因表达数据的标准流程是这样:
- Differnentail expression analysis 想了解转录水平不同处理的差别背后的原因
- Gene enrichment analysis (GO/KEGG) 得到差异基因,但是它们是干嘛的不知道,需要注释一下
但是有个弊端,它只能两两比较(如:感染与未感染),然后得到的结果也只是知道哪些上调哪些下调,是一个宏观的结论
使用Co-expression network 共表达网络可以分析多个处理的基因表达数据(例如:不同时间段处理),还能推断未知基因产物的功能、检测sub-groups
1.3 Network inference and reverse engineering
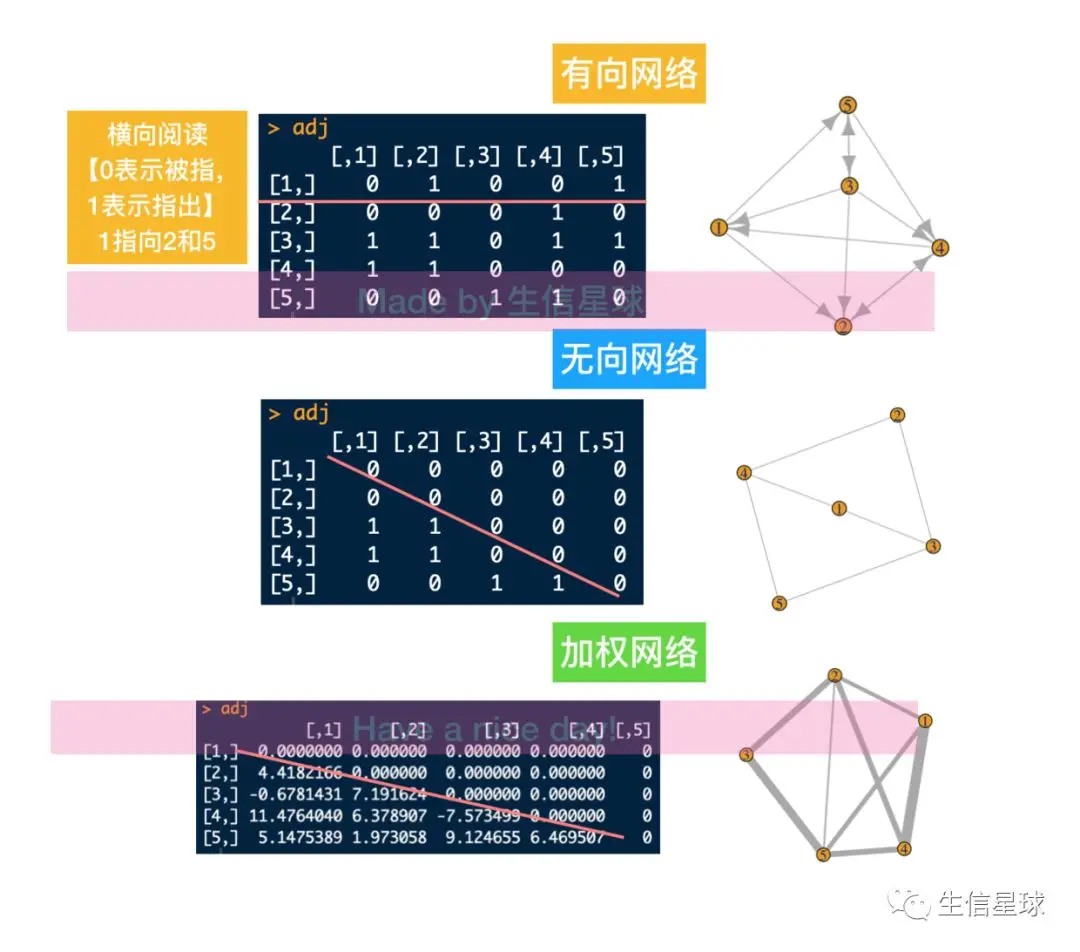
利用网络进行推断:可以使用表达量数据、已知的转录因子、ChIP-ChIP或ChIP-seq、时间序列等,因为网络是有向、交叉 的,所以可以判断许多的关系信息
说到网络,就要看一下有向和无向网络:
install.packages("igraph")
library(igraph)
set.seed(1)
# 先构建一个有向网络临近矩阵
# create a 5x5 adjacency matrix
adj <- matrix(sample(c(0,1), 25, replace = T), nrow = 5)
# set diagonal to zero (no self-loops)
diag(adj) <- 0
# plotting with igraph
g <- graph.adjacency(adj, mode = "directed")
plot(g)
#再构建一个无向网络
adj[upper.tri(adj)] <- 0 #就是取矩阵的下半部分
g <- graph.adjacency(adj, mode = "undirected")
plot(g)
#再构建加权网络 weighted network
set.seed(1)
adj <- matrix(rnorm(25, mean = 3.5, sd = 5), nrow = 5)
adj[upper.tri(adj, diag = TRUE)] <- 0
# note that igraph ignores edges with negative weights
g <- graph.adjacency(adj, mode = "undirected", weighted = T)
plot(g, edge.width = E(g)$weight)
构建共表达网络的关键步骤:
数据预处理 Data pre-processing
数据转换、过滤或者均一化都会影响下游的分析
选择数据: 如果想了解整体调控关系,可以选择全部样本,因为它们中间会存在不同的扰动,更能体现共表达; 也可以选择和某种表型相关的样本
过滤低表达基因 Filter low count genes
过滤低相关性/非差异基因 Filter low-variance/ non-DE genes :这是构建Robust性能更强的网络的关键一步
数据转换 Log2-CPM:将RNA数据转换成更像芯片数据的类型
标准化 Normalization
临近矩阵构建 Adjacency matrix construction
step1: 得到相似性矩阵
既然目标是构建共表达网络,那么就要找到这个“共”所在,即找到基因表达量的相似性,因此需要用到一些寻找基因间相似性的算法
基因共表达分析中最常用的两种相关系数
皮尔森 Pearson correlation,是线性相关系数,反映两个变量线性相关程度。如果两个基因的表达量呈线性关系(数学上,线性相关指的是直线相关,指数、幂函数、正弦函数等曲线相关不属于线性相关),那么两个基因表达量的就有显著的皮尔森相关系性。Pearson相关系数适用条件为两个变量间有线性关系、变量是连续变量、变量均符合正态分布。同一量纲数据可以选择Pearson,例如mRNA表达量数据 但在生物体内的许多调控关系,例如转录因子、小干扰RNA与靶基因,可能都是非线性关系,于是斯皮尔曼系数登场
斯皮尔曼 Spearman correlation ,是针对不同量纲计算的,比如两个通路看着相似,但其实单位不同,无法用pearson直接统计。无论两个变量的数据如何变化,符合什么样的分布,只关心每个数值在变量内的排列顺序。如果两个变量的对应值,在各组内的排序顺位是相同或类似的,则具有显著的相关性。
相关性|r|表明两变量间相关的程度,r为正表示正相关,为负表示负相关
step2 :根据相关性分组
- 方法一:不管正负,按照相关性绝对值分组
- 方法二:只将正相关的基因聚在一起
step3 : 将“假相关“去除
- 方法一:利用sigmoid 转换:1/(1+e^-x^ )
- 方法二:利用power转换x^n^
检测共表达模块 Network module detection
利用聚类树(反映数据的关系强弱)

纵坐标反映了相关性,数值越低越相似,其实也可以看作是相关性的反义指标,因此蓝色的那一坨就表现特别相关
下面主要介绍前期数据处理部分,数据处理好了,后面真正的WGCNA才能做好
2.1 Preparing RNA-seq data for network construction
- 数据质量控制
- 批次效应处理(ComBat/sva/RUVSeq))
- 归一化(Quantile normalization/TMM/etc.)
- Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments (Bullard, Purdom, Hansen, and Dudoit, 2010)
- A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis (Dillies, Rau, Aubert, Hennequet-Antier, Jeanmougin, Servant, Keime, Marot, Castel, Estelle, Guernec, Jagla, Jouneau, Laloe, Gall, Schaeffer, Crom, Guedj, and Jaffrezic, 2012)
- 关于RNA分析流程讨论
2.2 Prepare data and package
拿到一个数据,第一件事就是配置包
library('gplots') library('ggplot2') library('knitr') library('limma') library('reshape2') library('RColorBrewer') library('WGCNA')然后读取表型和表达矩阵
samples <- read.csv('sample_metadata.csv') raw_counts <- read.csv('count_table.csv', row.names=1) dim(raw_counts)大概的样子是这样:

加载注释信息
library('Homo.sapiens') #加载物种注释,这里以人为例
keytypes(Homo.sapiens) #查看注释包的内容,里面包含了各种的ID以及基因位置信息
# 比如要匹配Ensembl的原始ID到gene ID,并且显示染色体信息以及基因位置
gene_ids <- head(keys(Homo.sapiens, keytype='ENSEMBL'), 2)
select(Homo.sapiens, keytype='ENSEMBL', keys=gene_ids,
columns=c('ALIAS', 'TXCHROM', 'TXSTART', 'TXEND'))
# 就会得到类似下面的数据
## 'select()' returned 1:many mapping between keys and columns
## ENSEMBL ALIAS TXCHROM TXSTART TXEND
## 1 ENSG00000121410 A1B chr19 58858172 58864865
## 2 ENSG00000121410 A1B chr19 58859832 58874214
## 3 ENSG00000121410 ABG chr19 58858172 58864865
## 4 ENSG00000121410 ABG chr19 58859832 58874214
## 5 ENSG00000121410 GAB chr19 58858172 58864865
Sample check
分析之前,看看样本质量如何,可以用热图或聚类
# 用热图 # add a colorbar along the heatmap with sample condition { num_conditions <- nlevels(samples$condition) pal <- colorRampPalette(brewer.pal(num_conditions, "Set1"))(num_conditions) cond_colors <- pal[as.integer(samples$condition)] heatmap.2(cor(raw_counts), RowSideColors=cond_colors, trace='none', main='Sample correlations (raw)') } #用聚类 { sampleTree = hclust(dist(cor(raw_counts)), method = "average") pdf(file = "pre-sampleClustering.pdf", width = 12, height = 9) par(cex = 0.6) par(mar = c(0,4,2,0)) plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5, cex.axis = 1.5, cex.main = 2) dev.off() } # 如果发现跑偏的样本,除掉它 if(F){ abline(h = 20, col = "red") #画一条辅助线,h的值自定义 # 比如这里设置把高于20的切除 clust = cutreeStatic(sampleTree, cutHeight = 20, minSize = 10) table(clust) # 0代表切除的,1代表保留的 keepSamples = (clust==1) datExpr = datExpr0[keepSamples, ] }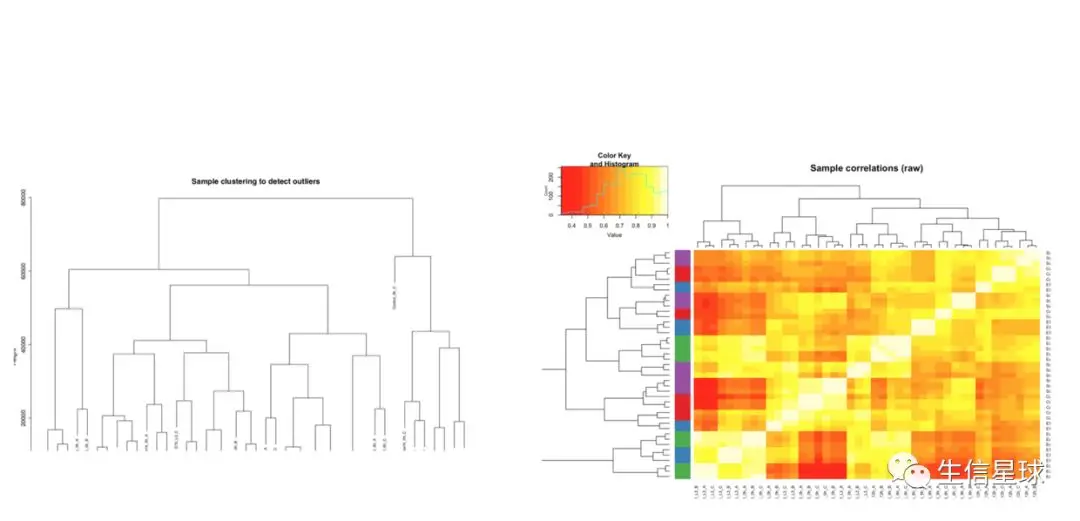
Low-count filtering 对样本质量满意后,我们只保留表达量丰富的基因,去除低表达量或者为0的基因
# 基因在每个样本中平均表达量为1就要被过滤 low_count_mask <- rowSums(raw_counts) < ncol(raw_counts) filt_raw_counts <- raw_counts[!low_count_mask,] sprintf("Removing %d low-count genes (%d remaining).", sum(low_count_mask), sum(!low_count_mask)) #结果会返回类似: ## [1] "Removing 6154 low-count genes (14802 remaining)."Log-transforming data
# 注意log使用要将真数+1 (真数就是这里的filt_raw_counts) log_counts <- log2(filt_raw_counts + 1) #然后画个图看看 x = melt(as.matrix(log_counts)) colnames(x) = c('gene_id', 'sample', 'value') ggplot(x, aes(x=value, color=sample)) + geom_density() #再画个热图 heatmap.2(cor(log_counts), RowSideColors=cond_colors, trace='none', main='Sample correlations (log2-transformed)')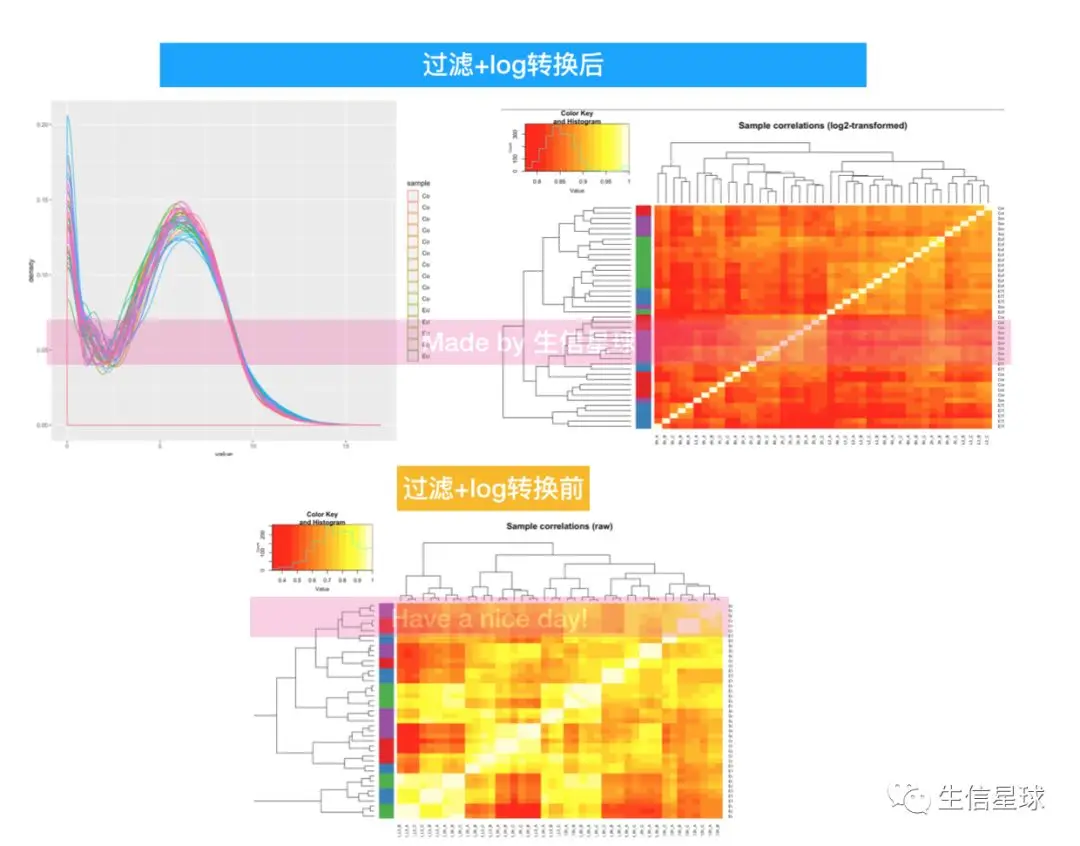
过滤并且log转换后,确实数据开始变好了
2.3 Remove non differentially-expressed genes
对于多个分组信息,需要生成几组两两组合的差异比较矩阵(取决于表型数据中的因子信息);并且方差不显著的基因就要去除
这里需要了解的有 quantile normalization、 voom
# 首先,去除方差为0的基因,因为这些基因没有表现出任何差别,还有可能对下面构建模型产生误导
log_counts <- log_counts[apply(log_counts, 1, var) > 0,]
# 构建design矩阵为差异分析作准备
mod <- model.matrix(~0+samples$tissue)
# 如果需要考虑其他分组信息,只需要加进来就好
# 例如:mod <- model.matrix(~0+samples$tissue+samples$cellline)
# 再改一下列名(因为默认是把对应的分组信息全部当成列名)
colnames(mod) <- levels(samples$tissue)
fit <- lmFit(log_counts, design=mod)
# 生成一系列的两两组合差异比较矩阵
condition_pairs <- t(combn(levels(samples$tissue), 2))
comparisons <- list()
for (i in 1:nrow(condition_pairs)) {
comparisons[[i]] <- as.character(condition_pairs[i,])
}
# 设置一个储存差异基因的向量
sig_genes <- c()
# 为每对差异比较矩阵都生成差异基因,并存储到sig_genes空向量中
for (conds in comparisons) {
# 这里conds如果中间有空格,就需要用make.names变成标准名
contrast_formula <- paste(conds, collapse=' - ')
# 然后就是标准limma流程
contrast_mat <- makeContrasts(contrasts=contrast_formula, levels=mod)
contrast_fit <- contrasts.fit(fit, contrast_mat)
eb <- eBayes(contrast_fit)
#p值可以自定义,这里设置的比较严格
sig_genes <- union(sig_genes,
rownames(topTable(eb, number=Inf, p.value=0.005)))
}
# 最后把差异不显著的基因去除,留下DEGs
log_counts <- log_counts[rownames(log_counts) %in% sig_genes,]