057-生信基础知识复习之测序
刘小泽写于18.11.25
每学习一遍之前的知识,总能从不同角度获取一些观点,然后扩增自己的知识库,这就是“知识迭代”
这次我也就是想到哪写到哪,把自己认为重要的内容梳理下,用Q&A的形式展示,没想到测序内容这么多,所以先当作第一部分吧
测序相关
Q1:生物体内的DNA的ATCG核苷酸是怎么转换成计算机识别模式的?
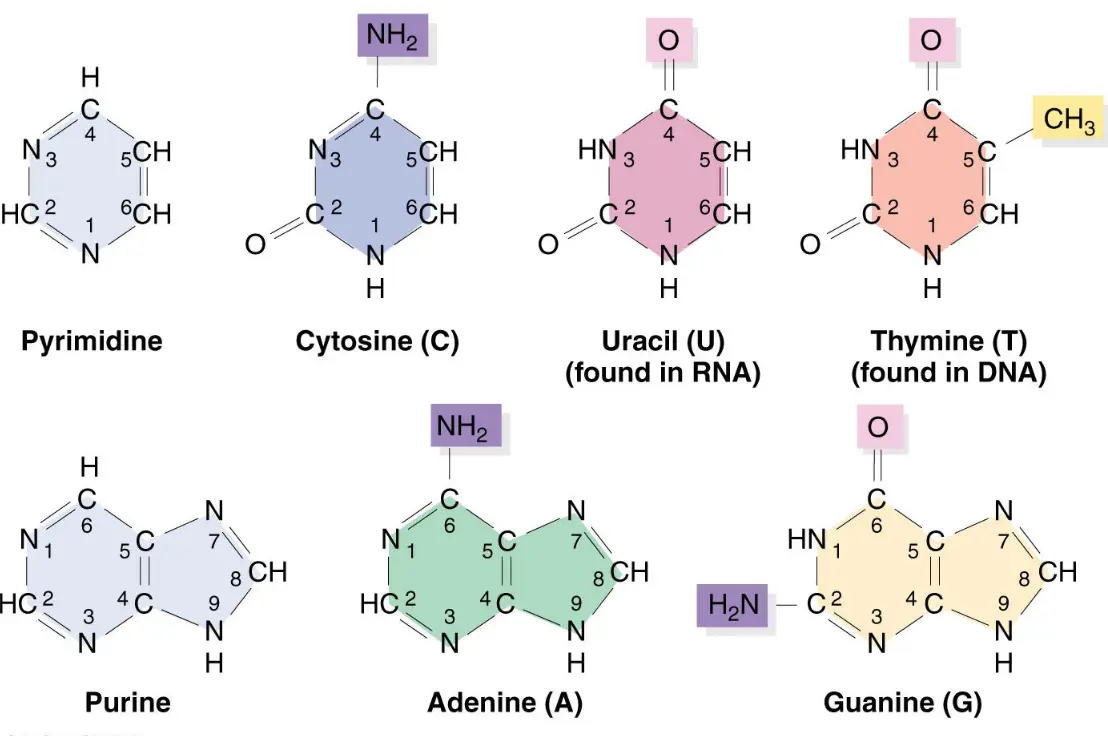
首先要区分
这是ATCG的化学结构,其中A、G结构类似,属于嘌呤类;C、T(U)结构类似,属于嘧啶类,我们测序的目的就是区别这几种结构。人类基因组中有30亿个碱基,要想区分其中的四类碱基,一个个分析结构肯定不靠谱,于是想到了使用颜色进行区分,也就是**“光信号”**方法
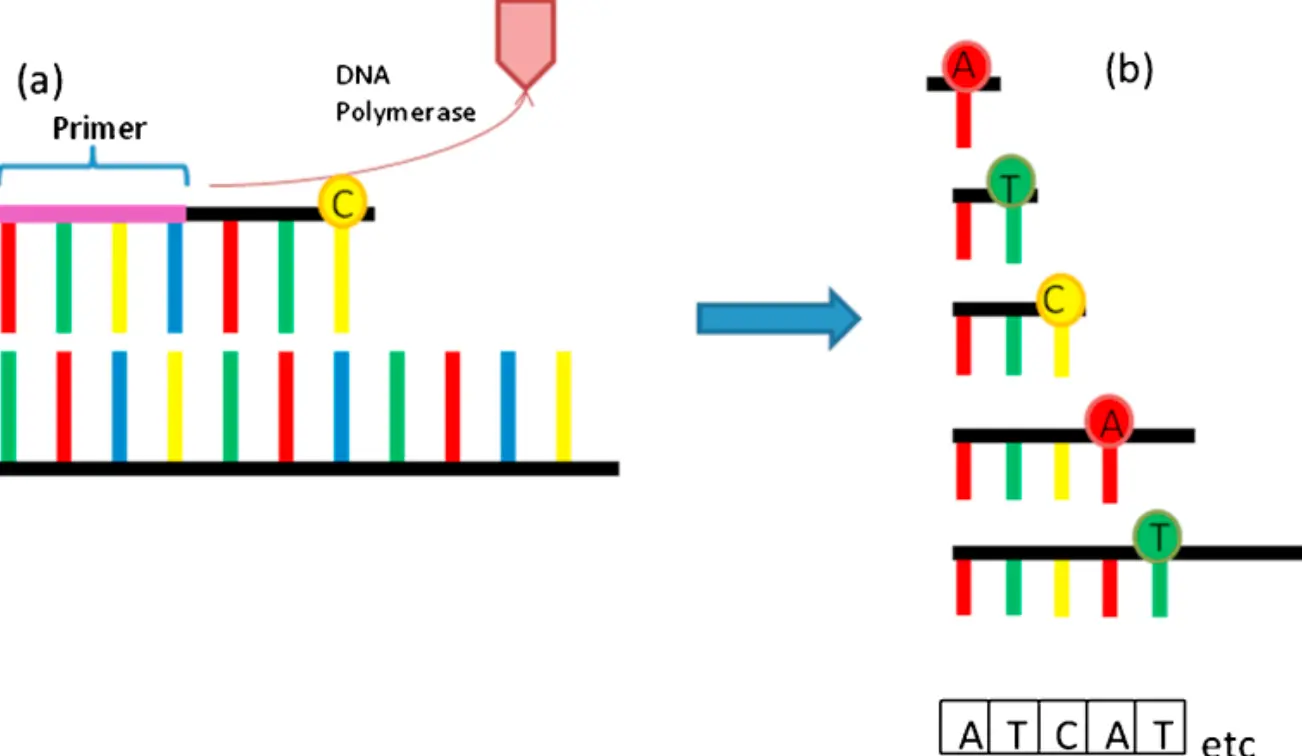
Sanger测序、Illumina、454等都是利用光信号。
光信号方法就是要让每个碱基带上颜色,就需要给每种碱基带上特定的荧光基团,荧光基团在测序仪激光作用下被激发,发出的光被照相机记录下来,然后荧光基团失效,接着加下一个碱基(利用了illumina的“可逆阻断终止技术”,简而言之,就是:在碱基3‘端加一个阻断基团,当聚合成功之后,就不能在继续3‘端加其他碱基了,这时利用激光捕获荧光信号,之后切掉荧光基团和阻断基团,让下一个带荧光的碱基继续进行)。
另外还可以用**“电信号”** (例如英国牛津纳米孔公司ONT的MinION测序仪以及18年被illumina以12亿美金收购的太平洋生物公司的SMRT技术)。基本思想就是:4种碱基结构不同,带的电荷就不同,在聚合的过程中,让它们通过电极,产生不同的电信号,利用电信号来区分【不过这种测序装置的灵敏度要求要比光信号更严格,才可以检测微小的电信号差别】
然后要规模
早起Sanger测序是利用了“末端终止技术”,这样准确但是效率比较低,因此illumina开发了“边合成边测序”,每合成一次就可以读取一个碱基,并且合成越长,测序读长就越长。这种技术就需要利用PCR进行大规模的扩增,但是我们知道,PCR技术收到酶活性的影响是有合成限制的,不可能无限扩增下去,illumina给出的解决方案就是:“双末端测序” ,就是正向测一段,反向再测一段,这样就在PCR循环一定的情况下,增加了测序读长
Q2:测序的基本流程是怎样的?
以常用的illumina二代测序为例,大体分为:建库、cluster、测序三步
**建库之前:**首先进行DNA样本的质量检测【1. 最好取单倍体(二倍体或多倍体有等位的杂合位点,测序时不容易分区哪种时物种真实存在的杂合位点,哪种时测序错误导致的假阳性 );2. DNA纯度要达到OD值要求(也就是DNA不能混杂蛋白质,对人和动物最好用红细胞提取DNA );3. DNA样本不能降解(因为测序时需要对样本进行随机打断,一般来讲,DNA越长,打断的随机性就越高,比如15K序列打断成500bp文库,这样打断的组合方式就很多,但1K的序列打断成500bp文库,组合方式就很少。如果降解成小片段,就无法进行随机打断);4. 测序量要够,能满足建库要求(测序的数据量是由加入的样本量决定的,并且为了重复需要保存备份)】
建库: 先理解“文库”(它是DNA片段的集合,将测序DNA随机打断就构成了DNA文库)。建库的过程可以想象成是硬盘格式化,出厂的硬盘(也就是未处理的样本DNA)直接插入计算机(测序仪)是不被识别的,需要分区挂载(也就是检测、打断、加接头等操作)后才可以。
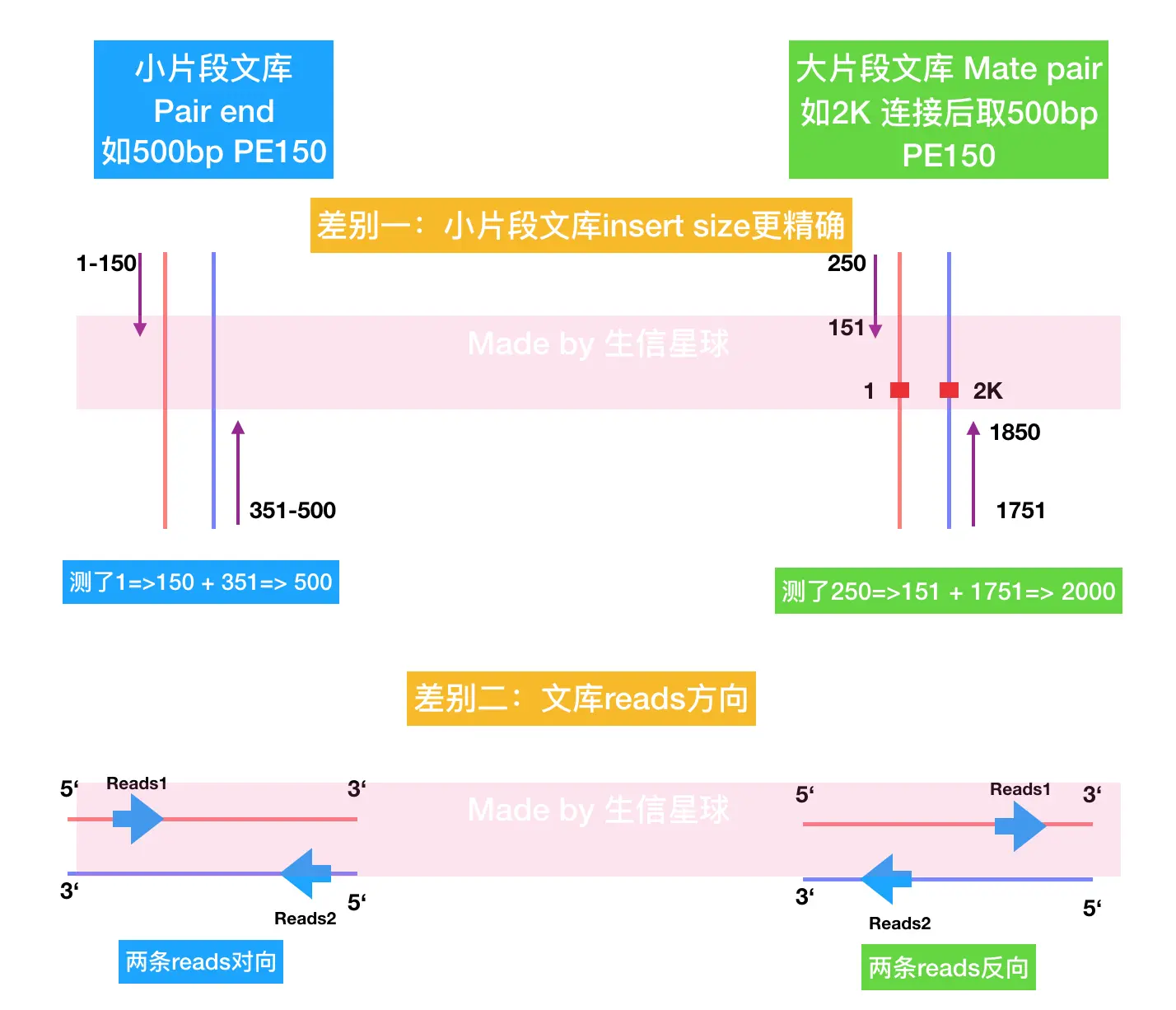
第一步:随机打断 这时的DNA是一条很长的片段(比如几百K的片段),使用机械法、酶接法、超声波(常用),然后设置打断的大小如500bp,然后这个长DNA就会断成许多的500bp的短片段,就形成了500bp左右的文库(需要注意:这里的500bp表示大部分片段在500bp左右,但并非每条片段都正正好好是500bp,可以存在400bp或者700bp)【常见的文库还有170bp文库、350bp文库、800bp、2K、5K、6K等】一般**1K以下属于小片段文库,1K以上是大片段文库。**文库大小的值又叫作“插入片段长度 Insert size”(这个值很重要,在序列拼接、短序列比对中会经常用到)
第二步:电泳 将一定范围内的DNA进行回收,如果要500bp文库,可以回收300-800bp的胶
第三步:3‘加A 在3‘加上一个A碱基,将原来平末端变成粘性末端,更容易在后续过程中连接引物和接头
第四步:加index标签 这是一个6-8bp的碱基片段,用于区分不同的测序样本。现在测序一条lane 能产生30G以上的数据,但是有的物种DNA没这么大,为了减少机器空转,可以将不同物种的DNA混合起来进行测序,但是下机后如何区分提交给客户呢?这就需要利用标签
第五步:加接头
加接头目的是将测序片段固定(或者叫“种”,种花的zhong)在flowcell的lane上,防止被测序的液体冲走。接头包括:P7接头、P5接头,其中P7接头和lane上的一样,P5和lane上互补,这样的目的是为了后面的桥式扩增
cluster: 这个词是“簇”的意思,也就是富集序列。因为测序需要荧光信号,如果仅对一条序列进行检测,那么信号很弱,识别错误率会很高,如果对一簇相同的序列同时检测某个位点的光信号,那么准确度就大大提高 这个过程就是“桥式PCR”,原来一条链以指数增长成为一簇“克隆”
序列条数x测序读长=一次测序量测序:加入dNTP(与桥式PCR中加的普通dNTP不同,它的3‘被叠氮基团修饰)、聚合酶。先从cluster同一条链的第一个碱基开始测,再到第n个碱基,直到测完整条链,得到reads1【注意这里才正式出现了reads这个名词,意思就是:一个碱基一个碱基读取】 测完reads1,加入碱性溶液将刚才测序完的链解链冲掉,加再入第二种测序引物,正好reads2的测序引物结合位点在index序列旁,先读取6-8个碱基测得index序列; 然后合成原来测序链的互补链,并切除原来测序链,还是按照原来方法测的reads2
测序完成后并没有得到想要的ATGC碱基顺序,而是一堆照片,下面就是图像处理转换成有颜色的光点文件(二进制BCL文件,即basecalling),其中包括测序仪编号、run序号、lane序号、tile号、X/Y坐标、index、reads1/2、碱基序列、质量序列、是否通过质量过滤(1为通过;0表示质量差)
Q3:已经知道文库有大片段文库(1K以上)和小片段文库,文库多大就能测多大吗?另外测序是双端测150bp左右,那么中间还有一部分没有测到,这样会不会有问题?基因组上的这部分区域会被忽略吗?
首先,不管构建多大的文库,测序得到的都是两端很短的序列(比如双端150就是得到两个150bp的reads);
其次中间测不通并不会有问题,而且是非常正常的现象!因为文库构建是随机打断过程,所以即使第一条片段中间没有被测到也没关系,后面的其他片段一定能测到这中间的部分(因为一次测序过程会产生成百上千万条reads,而基因组就那么大)
另外,你可能会想:既然只能测两端很短的一部分,那么小片段和大片段文库的区别在哪?反正都测不完。其实,大片段文库的目的,除了得到序列以外,更重要的是,为了获取片段的坐标距离(即两条reads之间的物理距离关系,将会为序列拼接和基因组结构变异检测提供帮助) 。当然,目前大片段文库还有一些问题,比如现在PCR手段不能扩增太长的片段,另外我们只能测两侧的很短的片段,那么中间合成出来却不能测,造成了浪费!
但是这些问题illumina给出了大片段文库的解决办法:
在随机打断序列后,大片段比小片段文库多了一个环化处理,经过末端修复,再将一个线性长片段头部进行生物素标记,再进行环化(即:把片段首尾连接成一个环)【我们现在知道了:小片段文库是pair end; 大片段文库是mate pair】 曾经我也是为这两个概念搞的头晕转向🥺
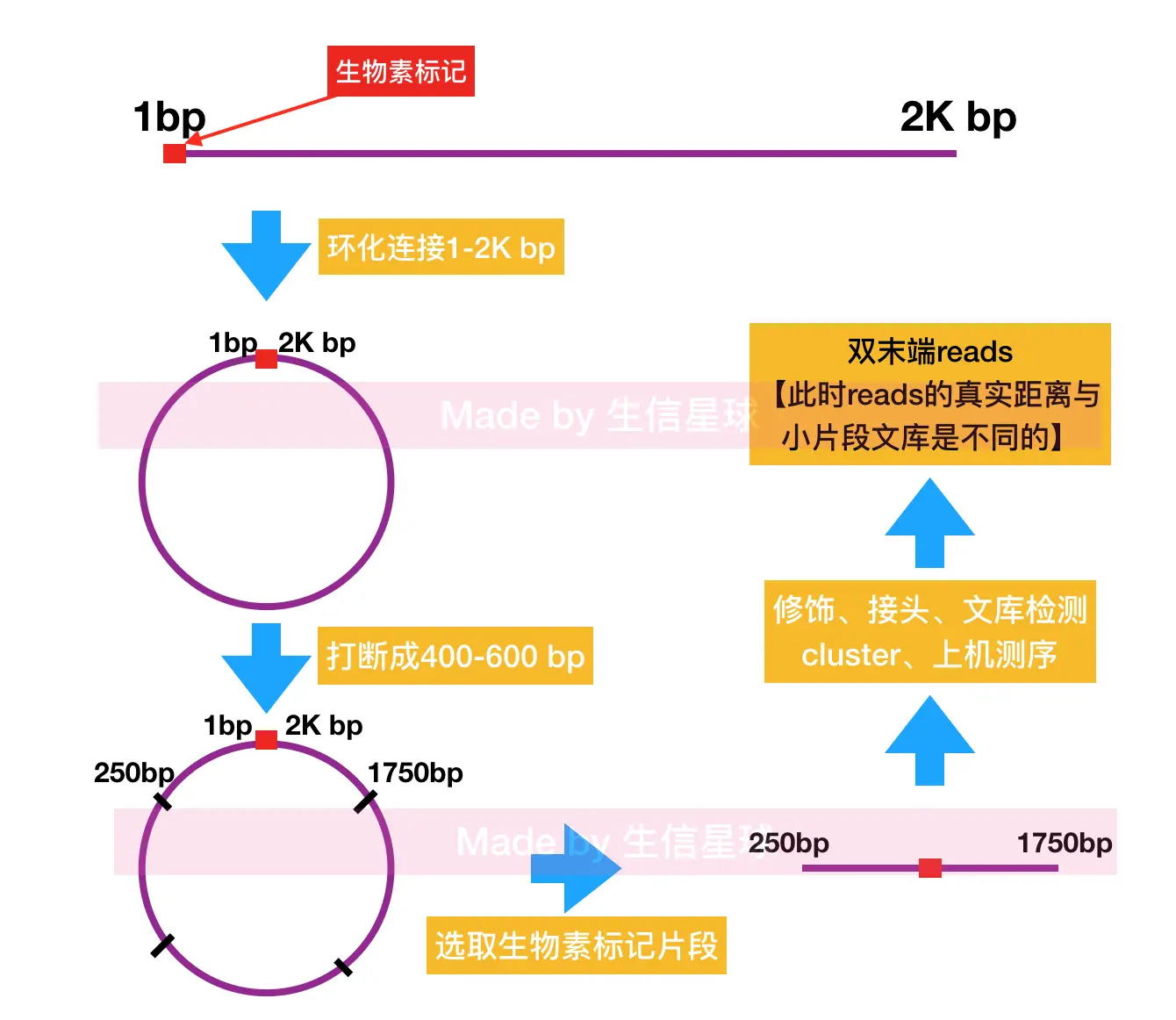
关于大小文库的差别:
Q4:为什么不能测完整的基因组?
理想的情况是:基因组有多大,我们就能测多大
但事实是:我们提取的DNA就不是完整的一整条,而是断成许多片段,比如10M基因组提取出来,可能也就剩一堆几百K的片段。现在可以做的就是对这些几百K的片段随机打断测序;另外,目前二代测序基本都依赖PCR扩增,因此限制了读长
Q5: 目前市面上一二三代测序并存,怎么选择?
存在即合理,因为没有任何一种测序技术能胜任任何工作,才会出现现在的局面。对于选择困难症患者来讲,一般可以从测序读长、通量、准确性、价格角度考虑
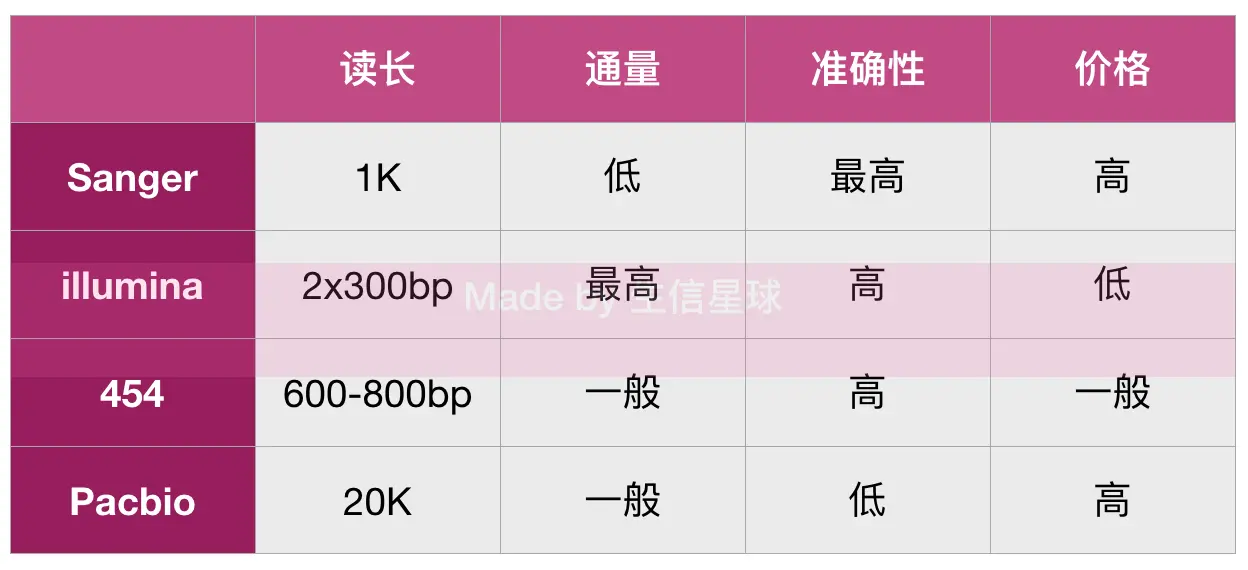
所以也能理解,为什么illumina是目前的龙头,另外收购Pacbio后它在三代的市场又可以一展拳脚了
Q6: GC bias是什么意思?
基因组正常的GC含量是35-65%,如果小于35%或者大于65%就属于异常。我们知道AT是2个氢键连接,而GC是3个氢键,因此如果GC含量太高,在PCR过程中解链需要的能量更高,导致模版链更难打开,默认的温度下,DNA模版变性不完全;另外PCR产物难易结合到模版,DNA聚合酶也难以延伸,结果就是出现非特异性条带,不容易被扩增,因此也无从谈及测序,最后基因组覆盖不均匀,丢失部分信息
对于这样的样品,可以构建PCR-free文库 ,但需要更多样本量
Q7: 关于读长、插入片段大小的选择
我们知道了小片段测序文库中有多种规格可供选择,如:170bp、350bp、500bp、700bp等。读长的话,在保证准确性前提下,越长越好,有利于序列拼接。例如Miseq可以实现PE 300bp的读长,如果选择500bp文库和Miseq PE300(效果可以和454差不多了),那么中间就会有100bp重叠区域,发生了所谓的“片段测通”,可以利用这个区域将两个reads拼接起来,形成更长的序列。
文库大小需要和reads读长相协调,对于较短的测序片段,文库不能过大;对于De novo 拼接,可以先使用小片段文库,然后转为大片段文库,并逐级增加文库大小如2K、6K、10K、20K等【目的就是合理使用重叠区域进行逐级拼接】
Q8:为什么小片段文库如500bp需要两端分别测,而不能一次测通500bp呢?
利用PCR反应是可以实现的,就是一直扩增,把500bp全部测出来。不能这么做的一个因素是:PCR中DNA聚合酶的活性会下降,因此测序错误率会随着测序长度增加而增加 ;另外一个因素就是Phasing,按说cluster中所有片段都要保持同步,第一次大家都加第一个碱基,第二次都加第二个碱基… 但实际上,总有几个走的快或者走的慢(一次加两个碱基或者这一次一个碱基也没加),这些“离群”的碱基出来的荧光值就会带给整体干扰
Q9:不同的测序,不同的操作?
对于全基因组测序,就是按上面的测序步骤就好;
对于转录组测序,就需要考虑RNA反转录的问题,那么是先反转录再打断还是先打断后反转录呢? 其实比较高效的方法是:先反转录后打断。一般转录本比较短(小于2K),那么选择文库时就不能太大(比如,不能选800bp文库,因为2K的序列,打断成800,随机性不是很好),可以考虑小一些的文库(300左右)
另外还有很多测序类型:外显子组、甲基化、小RNA、宏基因组等