055-一起来爬聚类树!
刘小泽写于18.11.21
聚类树是个有意思的东西,可以十分直观地看到每个物种的亲缘关系 之前没有涉及到相关的分析,相关的工具只是听说过大家都在用的MEGA,另外还有Y叔的ggtree。很好奇但是一直没时间学,但是昨天被问到一个关于Bayes建树的问题,于是我想试试。这次先简单介绍下贝叶斯方法,从难入易循序渐进
聚类树什么用?
我们经常听到:系统发育树、系统进化树、系统发生树,其实都是为了推测物种进化机制、预测机制背后的关键作用位置
概率名称
- 先验概率:基于背景知识、历史数据等经验,在事件发生前预先判断的概率
- 后验概率(posterior probability):贝叶斯统计中,一个随机事件或者一个不确定事件的后验概率是在考虑和给出相关证据或数据后所得到的条件概率(来自:维基百科)它是基于先验概率求得的反向条件概率
建树方法
基于距离的方法包括:UPGMA(现在很少用了)、ME(Minimum Evolution 最小进化法)、NJ(Neighbor-Joining 邻接法)
基于特征:MP(Maximum parsimony 最大简约法)、ML(Maximum likelihood 最大似然法)、BI(Bayesian Inference贝叶斯法)
建树完成,一般需要用bootstrap(自展率)或者Posterior probability(后验概率)来评估
贝叶斯建树
这种方法是最准确,但同时也最慢【貌似这是软件的通病,比如比对软件STAR也是如此】
软件安装
软件的官网在:https://nbisweden.github.io/MrBayes/download.html
提供了pdf的官方教程【后台回复“mb”获取】
软件安装可以选择三大系统平台,但是我还是比较推荐linux,能用服务器就用服务器运行,一是节省时间,二是减少自己电脑消耗。
安装也很简单,直接用conda安装就好,为了避免软件安装过程中出现一些潜在的冲突问题,可以新建一个conda 环境
conda create -n mrbayes
source activate mrbayes
conda install -y mrbayes
快速开始
快速教程分为四部分:读取Nexus文件、设置进化模型、运行程序、归纳样本
第一部分:先下载好一个测试文件
mkdir ~/mrbayes && cd ~/mrbayes wget http://ib.berkeley.edu/courses/ib200b/labs/primates.nex然后输入
mb就会运行mrbayes命令【需要注意的是,每一个mb的命令都是以MrBayes >开头】使用命令
execute primates.nex就把数据读进来(如果你的文件不在当前操作目录,最好使用文件全路径表示)
第二部分:在
MrBayes >后面,输入lset nst=6 rates=invgamma这是将进化模型设置成GTR(General Time Reversible)模型,包含了变异位点的gamma分布率和非变异位点的比率【只是针对此处的演示DNA数据】
如果数据不是DNA或者RNA,或者想调用(invoke)其他模型,又或者不想用默认参数,需要用到后面进阶的知识
第三部分:在
MrBayes >后面,输入mcmc ngen=20000 samplefreq=100 printfreq=100 diagnfreq=1000这段代码的结果会从后验概率分布中得到至少200个sample,然后每1000 generation计算一次结果;如果数据集比较大时,可以将样本频率调低一些,运行的长度更长。【默认的设置是sample和print的frequency是500,diagnfreq是5000,ngen为1,000,000】每一行的末尾是估算运行剩余时间
运行结束后,会给出一个结果:average standard deviation of split frequencies。如果得到设置的20000 generations后,split frequency 是小于0.01的,就可以停止运行了(输入
no);否则继续运行,直到这个值变成小于0.01。当然,0.01是一个比较严格的阈值,如果只对树中构建的比较好的部分感兴趣,那么split frequency小于0.05也是可以的第四部分:输入
sump就是summary of parameter,包括了mean, mode, 95% HPD (Highest Posterior Density) interval需要注意的是,PSRF (Potential Scale Reduction Factor) 应该在所有参数中接近1;如果差别比较大,需要设置分析长度再长一些

接下来输入
sumt,会根据后验概率对每个分支生成一个进化分支图,也会根据平均分枝长度生成系统发育图,这两个树图可以输出成被FigTree、TreeView、Mesquite等画树软件识别的文件
详细点的解读
第一步 读取数据
数据格式需要是Nexus,包含比对好的核苷酸或氨基酸序列、形态学数据、限制性位点数据或者这四种数据的混合。【关于Nexus格式的介绍:http://wiki.christophchamp.com/index.php?title=NEXUS_file_format】
Nexus数据中,不同的数据支持的字符是规定好的,例如
- DNA数据
A, C, G, T, R, Y, M, K, S, W, H, B, V, D, N
RNA数据
A, C, G, U, R, Y, M, K, S, W, H, B, V, D, N蛋白数据
A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V, X限制性位点数据
0, 1形态学数据
0, 1, 2, 3, 4, 5, 6, 5, 7, 8, 9
如果出现某些模糊的位点,可以用圆括号或者花括号注释,例如
# 不例如清楚氨基酸是A还是F
(AF), (A,F), {AF}, {A,F}
如果使用自己的真实数据,在导入数据后,需要将自己的数据调成和示例一样的格式,就是类似这种。自己需要修改
ntax(总共要聚类的sample数),nchar(每个sample的序列字符数),datatype(DNA/RNA/protein等)。如果有missing或者gap,需要说明他们用什么字符表示(比如:missing=*gap=-)

使用execute或者exe + 文件名(有路径的需要写路径) 来加载.nex文件
第二步 指定模型【重要的部分】
在加载数据以后,我们可以使用showmodel来看看针对自己的数据类型,有哪些模型可选,然后需要用到的命令:lset定义模型结构,prset定义模型参数的先验概率分布
lset:使用help lset 看一下怎么设置lsetlset <parameter>=<option> ... <parameter>=<option>
还有一个表格,表示了参数及当前设置
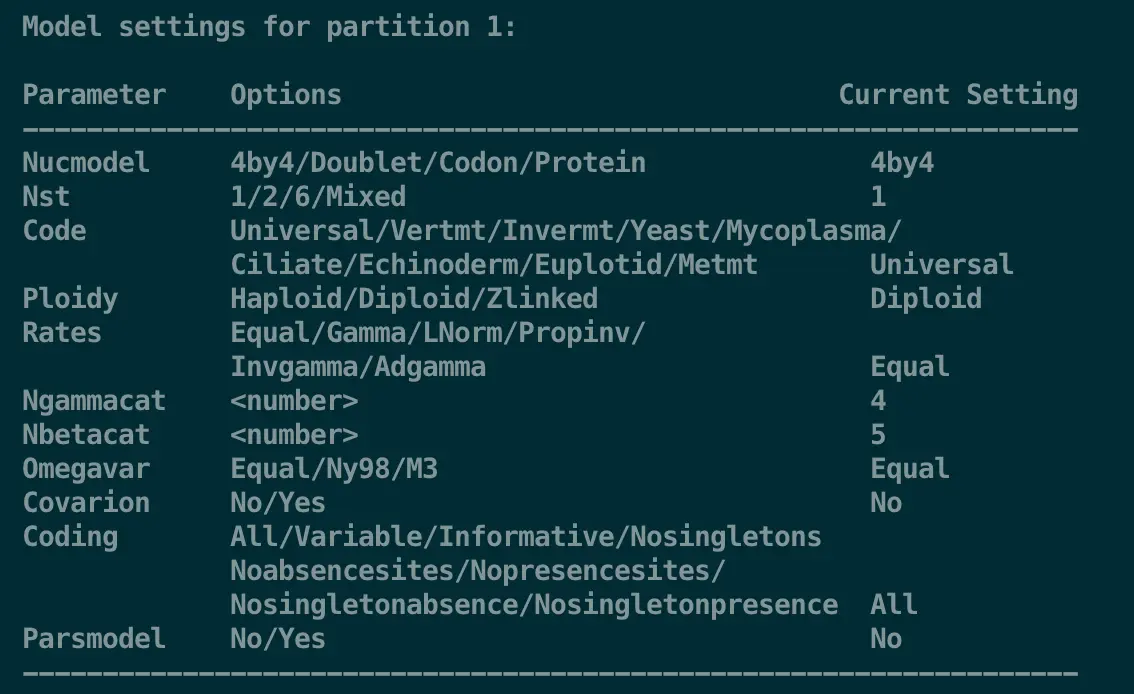
行首:Model setting for partition 1,表示的是你要比较的序列都是同一类型,如果不同类型就会分成不同的partition;
第一个参数 Nucmodel: 设置核苷酸替换类型。默认是4by4(意思是核苷酸的四种形式ACGT/U);Doublet是核糖体DNA(即编码rRNA的序列)的成对茎区 [paired stem regions of ribosomal DNA]; Codon是利用密码子分析DNA序列;Protein也就是DNA转变的氨基酸序列;
第二个参数Nst:用数字设置替换的种类。1表示所有替换率都一样(比如JC69 or F81 model);2表示所有转换和颠换的比率由一些差别(比如K80 or HKY85 model);6允许所有的替换率存在差别(如GTR model);mixed表示在所有可能的可逆替换模型中进行“马尔科夫链”抽样,并组合出不同的模型;
第三个参数Code:只有当Nocmodel设置为Codon时才有效,默认就是全部密码子;
第四个参数Ploidy:设置染色体倍数,“Haploid”, “Diploid” or “Zlinked”;
第五个参数Rates: 设置模型的位点变异率。默认是equal(即所有位点没有变异);gamma表示位点变异符合gamma分布;lnorm表示正态分布的对数;propinv表示变异位点的分布是常数;invgamma表示变异位点的分布是常数,但是其他位点分布是gamma分布;
第六、七个参数Ngammacat 、Nbetacat一般用默认值4、5就好(大多数情况下,Ngammacat取4个rate categories就够了,增加rate categories数量可以增加准确度,但同时速度会变慢,4可以理解为一个折中的值);
第三步 设置模型先验值及检查模型
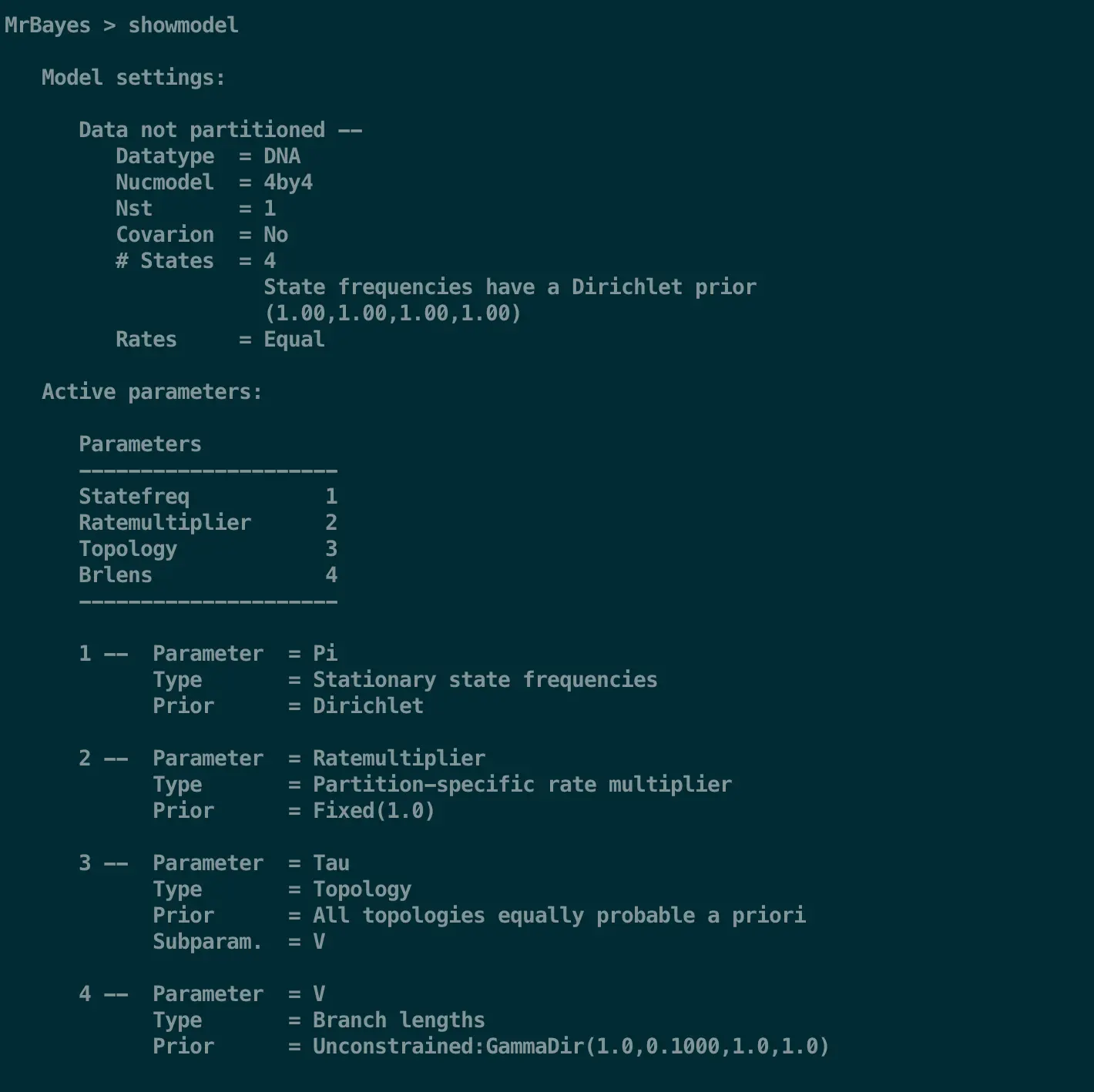
有六种参数类型:拓扑(the topology), 分枝长度(the branch lengths), 4个stationary frequencies of the nucleotides, 6个different nucleotide substitution rates, the proportion of invariable sites 以及 the shape parameter of the gamma distribution of rate variation
默认值可以应对绝大多数分析,使用help prset可以看到设置
使用shomodel会看到目前模型的参数设置
第四步 准备分析
在运行mcmc 之前,可以用help mcmc看下
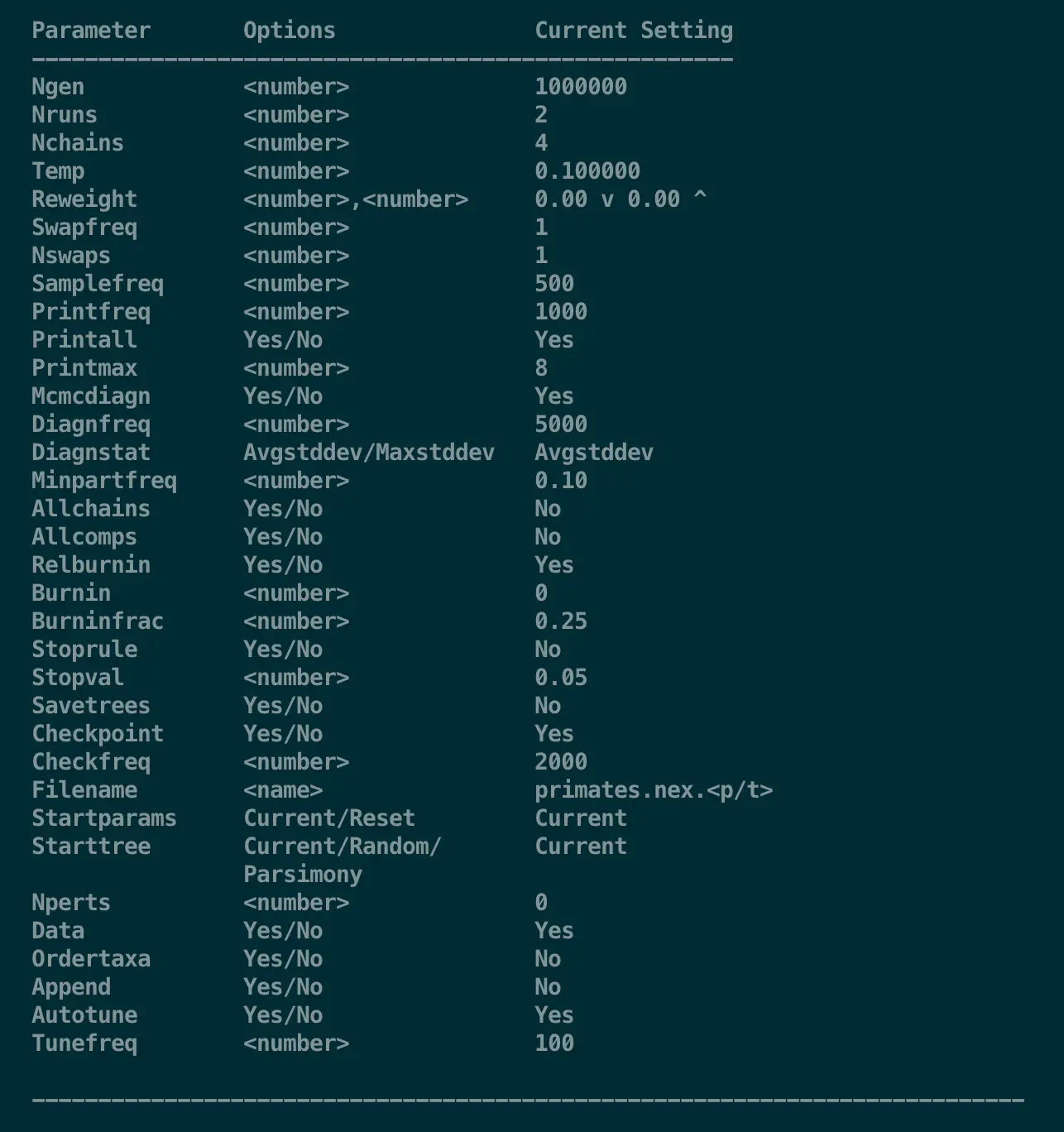
- Ngen:generation的数量,可以先设置个小一点的值确保整个流程是正确的,然后还可以根据这个结果估算更大的Ngen使用时间。这里的测试数据我们使用20000即可,但是大数据集可以适当调小一些。使用
mcmcp ngen=20000进行调整,然后再用help mcmc确认 - Diagnfreq:每隔多少generation进行一次评估,对大数据集来讲设为5000比较合适,这里测试的12个样本设置1000比较合适
mcmcp diagnfreq=1000这个结果会保存在.mcmc文件中。每次评估结果出来,都会从马尔可夫链开头根据burnin(固定的要被丢弃的样本数量)或者burninfrac(样本百分比)丢弃样本。默认情况下,cold chain的前25%会被丢弃(通过relburnin=yes and burninfrac=0.25设置) - Nchains:默认情况下为4(3个heated chains,1个cold chain)一般来讲,增加3个heated chains对复杂的大型数据集有帮助,当然链数越多对内存消耗越大。在服务器上,可以使用MPI version的mrbayes软件来分配链给不同的CPU,提高运行速度
- Samplefreq: 链被取样的频率,默认是链上每隔500单位被取样一次。小数据增加频率,也就是减小数值比如测试的数据中12样本只需要设置成
mcmcp samplefreq=100;大的数据集可以适当增加数值。当链被取样结束后,参数信息被储存在.p的文件中(tab分割); 拓扑和分枝长度被输出到.t文件(这些是Nexus tree file,可以导入TreeView) - Printfreq: 就是输出到屏幕的结果间隔,默认是500
第五步 开始分析
输入mcmc ,首先看到这样一个表格,包括了需要用到的方法及占比
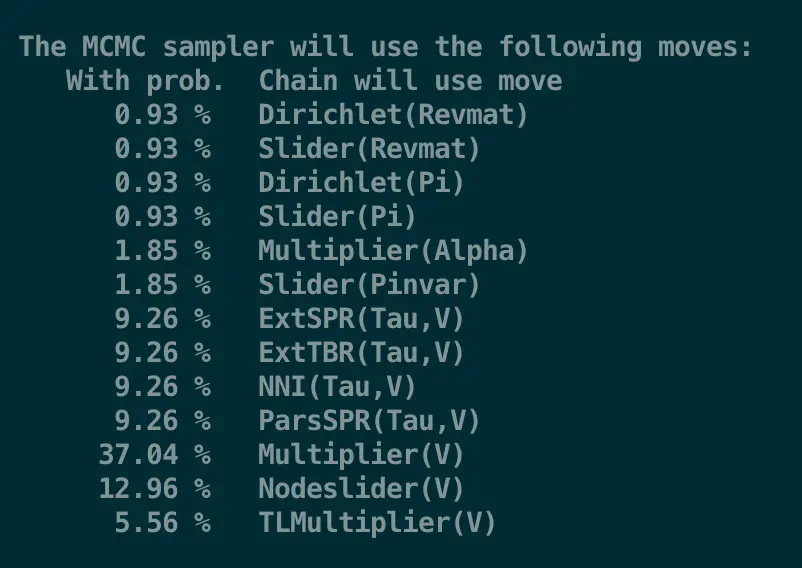
然后就开始运行,就像这样
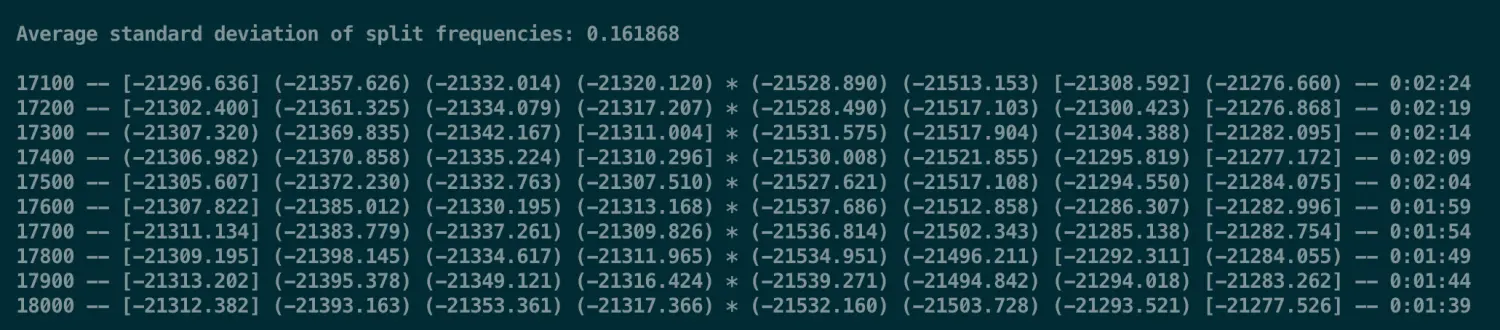
第一列是 generation number,后面8列负数代表内存的物理位置
2-5列是run1的数据(之前看到Nruns=2,说明有两个完全独立并同时进行的run),一列的每行对应一个chain。
最后一列是预计剩余时间
什么时候停止呢?
当程序运行到设置的Ngen时,会问你要不要继续,并给出目前已经计算的结果Average standard deviation of split frequencies。一般来讲这个数小于0.01就可以退出了。但是在数据集比较大时,并不是很理想,下降的很慢,而且开始还有时会上升,如果到了Ngen还离0.01很远,比如最后还是1点几,那么还需要增加generation。如果达到了0.01到0.05之间,那么基本可以结束。
第六步 总结
首先看下samplefreq生成的.p文件,不同的模型结果信息不同【这里以测试数据为例】

- 第一行:每个运行结果独立的ID号;
- 第二行:从左往右依次为: Gen (generation number); LnL (log likelihood of the cold chain); TL (total tree length/ the sum of all branch lengths); r(A<->C)等 (six GTR rate parameters); pi (four stationary nucleotide frequencies); alpha (shape parameter of the gamma distribution of rate variation); pinvar (proportion of invariable sites)
然后使用sump 得到一个图一个表。生成的表格中,主要看PSRF这一列数值是否接近1.0【1.00-1.02是最理想的情况,但很难做到】

tree和branch数据存储在.t 的文件中,也是Nexus格式
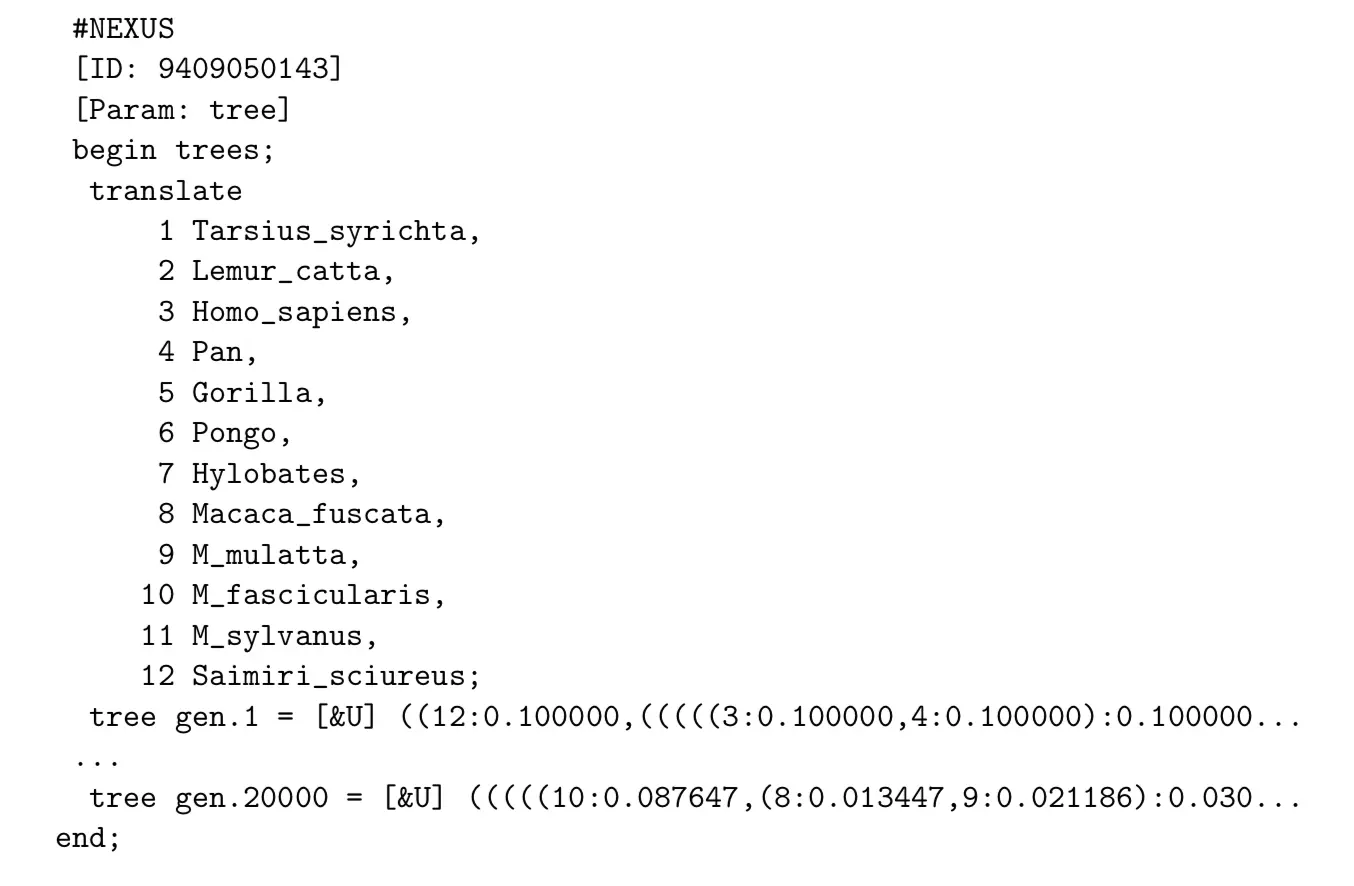
使用sumt relburnin =yes burninfrac = 0.25 ,直接返回的结果包括:两个统计表两个图
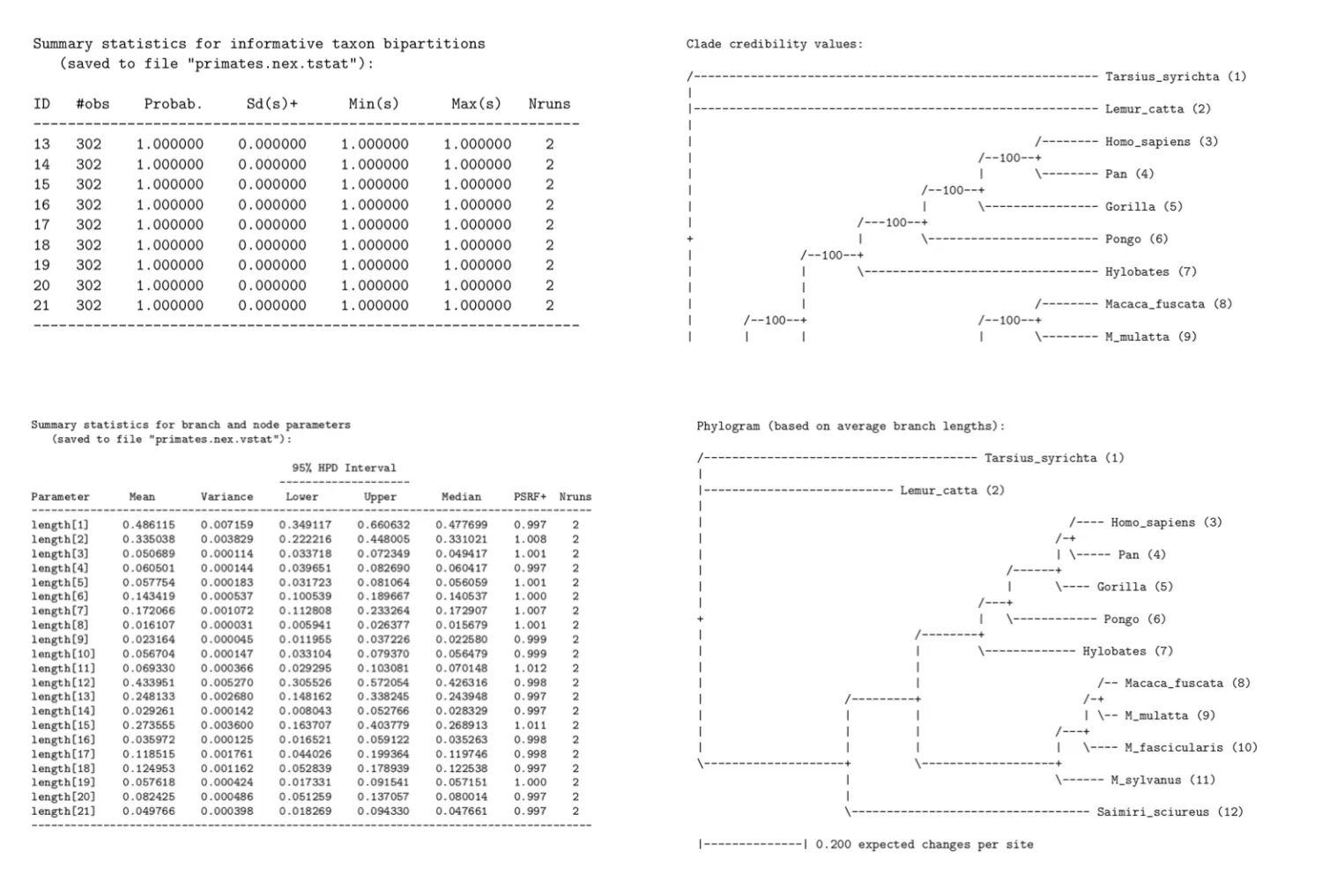
左上:
obs表示number of times the partition was sampledProbab表示probability of the partitionSd表示standard deviation of the partition frequencyMin/Max表示min and max of the standard deviationNruns表示number of runs左下:记录了length数据,其中length的编号对应左上图中ID号,依然关注PSRF列(Potential Scale Reduction Factor)
右上:clade credibility tree: probability of each partition or clade in the tree
右下:phylogram tree:branch lengths measured in expected substitutions per site
另外,sumt还会返回5个额外的文件
.parts文件包含了二分法分类的key值;
.tstat和.vstat 包含了partition statistics和branch length statistics
.con 包含了consensus trees,这个文件可以在FigTree中打开,展示一下后验概率以及每个枝的标准差【这个应该是比较有用的】
.trprobs包含了在mcmc搜索中找到的树,并用后验概率排序