053-Statquest之聚类分析
刘小泽写于18.11.17
今天写写从statquest上学到的关于聚类的一点知识
相关背景
聚类,又叫做Hierarchical clustering,经常和热图联系在一起展示
我们一般看到的热图是这样的:
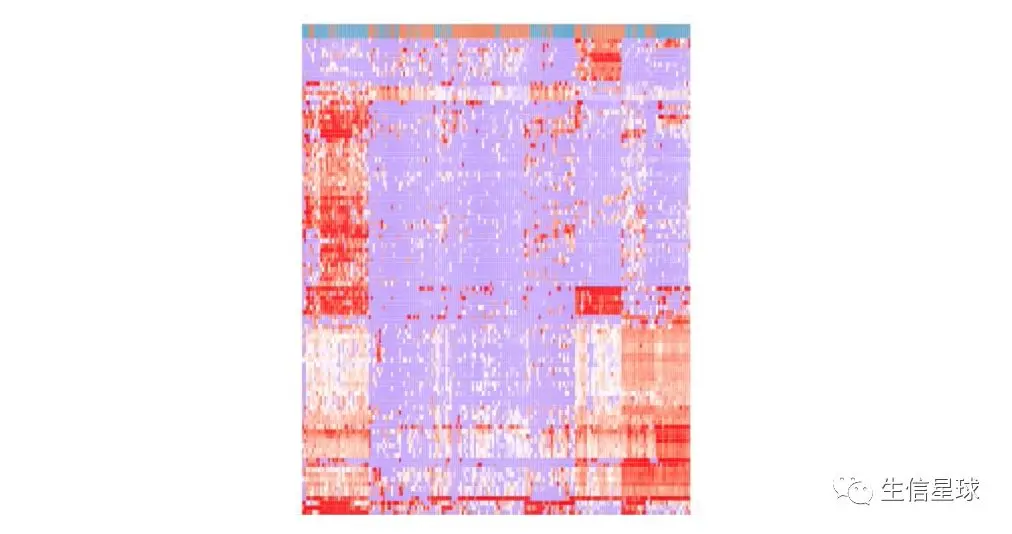
列为不同的样本,行为不同的基因测量值(比如表达量),不同的颜色又区分了大小(比如这张图中红色表示高表达基因,而蓝色/紫色表示低表达)。聚类分析的作用就是将行或者列根据相似程度排序,越相似排的越近,于是我们观察数据的相关性就方便多了

怎么做的【不涉及算法】
我认为这部分对于选择困难症用户就比较尴尬了,因为有时候真不知道应该怎么选
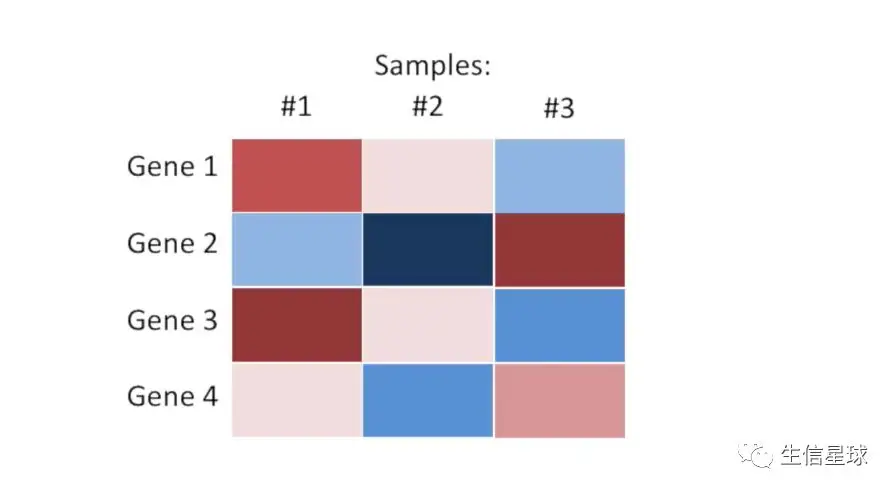
假设我们有四个基因,每个基因各测了在三个样本中的表达量,我们现在想对基因(也就是每一行)进行重排序
基因之间相互PK环节
首先看基因1,在三个样本中表达量分别是“褐色”、“粉色”、“蓝色”(这里先不考虑颜色深浅,只看大体的颜色划分)。基因2和基因1在三个样本中颜色差别还是很大的,但是基因3就和1更为相似 ;
第二步就是看基因2,找到和基因2最相似的基因,也就是基因4;
然后就是挨个基因都要找和它们最相似的一组;
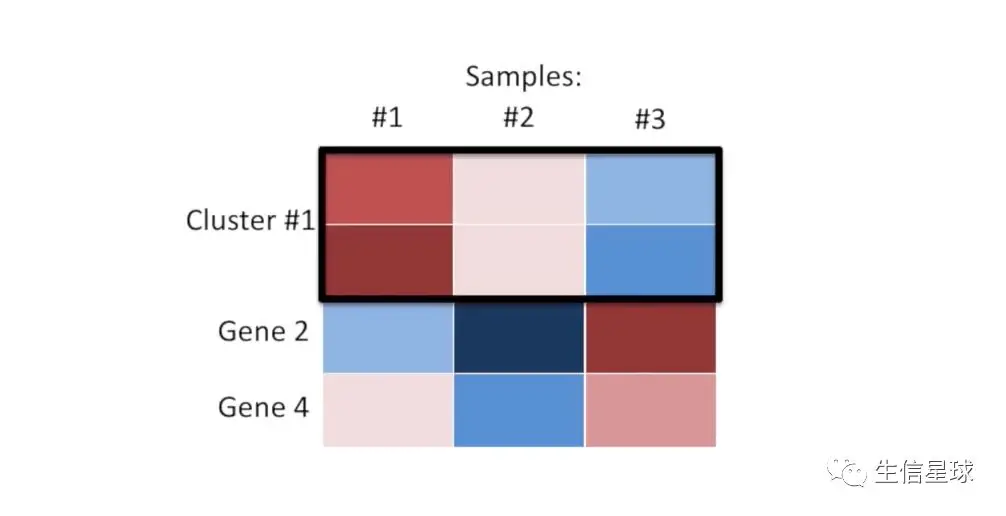
得到每个基因和它最相似的基因后,它们组成一组。我们再将这些组放在一起进行比较,看那一组基因是所有组中最相似的,挑出来放到一个cluster(簇)中
【这里,基因1和基因3这个组合就是所向披靡的,打败了其他的组合,因此它们被放在一起形成cluster1】,就像这样:
基因与cluster之间相互PK环节
上面获胜的基因1和3组成的cluster1将成为新的挑战对手,虽然这个cluster包含两个基因,但还是要再接下来的环节中当作一个基因看待,让剩下的基因(gene2和gene4)再与这个clustet“基因”比较,可以看到基因4与这个cluster“gene”的相似性比基因2要高;
然后第一个“基因”被比完了,接下来还是比第二个基因,看基因2和谁的相似性最大,结果看到基因2和基因4比基因2和基因“cluster”更相似
于是我们挑出了下一个cluster2(基因2和4)。。。
合并clusters
当把所有的基因都找到它们的归属cluster后,我们就可以把它们合并
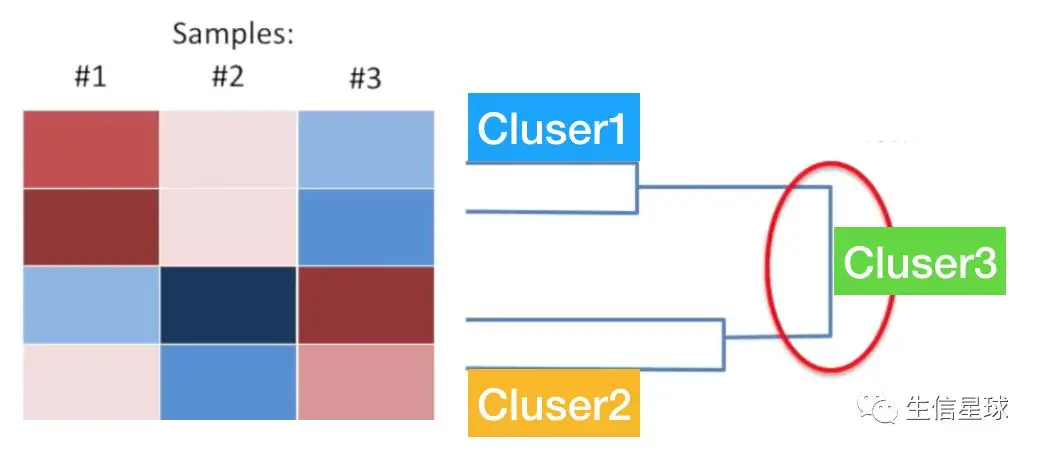
结合树状图dendrogram
分析完cluster后,需要用线条表示远近关系,之前的cluser1是第一个获得的,并且它其中的基因相似性是所有ckusetr中最强的。因此体现在树状图上就是cluster1的分枝最短;以此类推,cluster2的分支长度排第二位;而最后合得到的cluster,它的分支是最长的
算法部分
比较基因
我们的第一步是:找出和基因1最相似的基因,那么问题来了:如何定义相似性?
这个判定条件仁者见仁,但是大家都熟知的方法是:计算基因间Euclidian distance
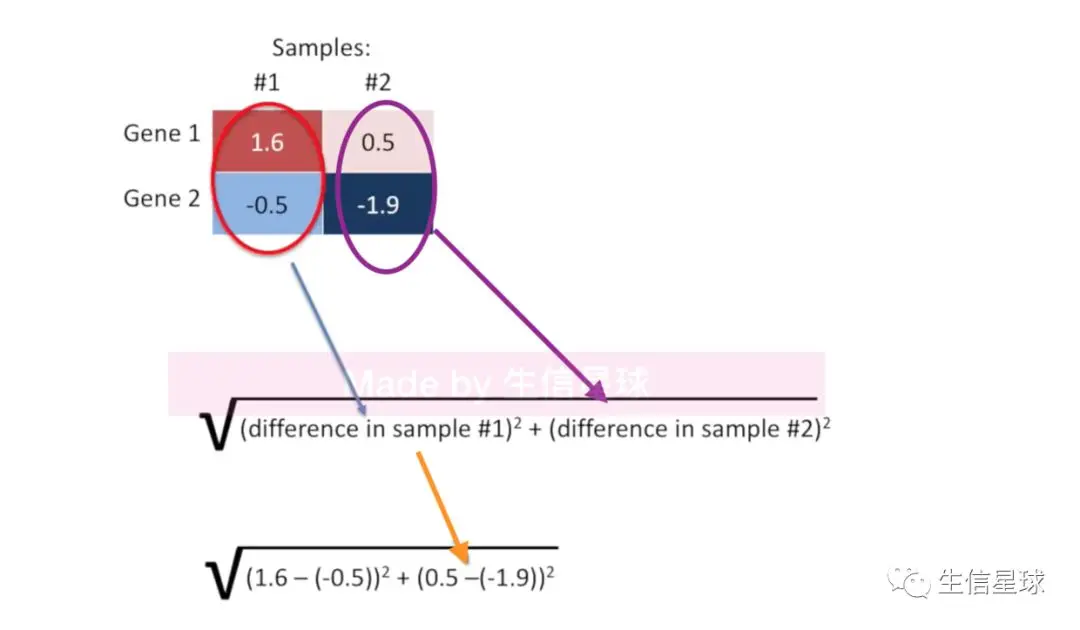
这里可以想象一个直角三角形,其中一个腰长度是:1.6-(-0.5)=2.1【表示样本1中基因1和基因2的距离】,另一条腰长度是0.5-(-1.9)=2.4【表示样本2中基因1和基因2的距离】,然后三角形的底就是上面👆公式计算得到的结果(3.2):表示了样本1和2中基因的总距离
这里只是用了两个样本,但是一般我们都是十几个甚至更多的样本,于是:我们只需要将上面公式进行拓展,在根号中增加到(difference in sample #n)^2^ 即可
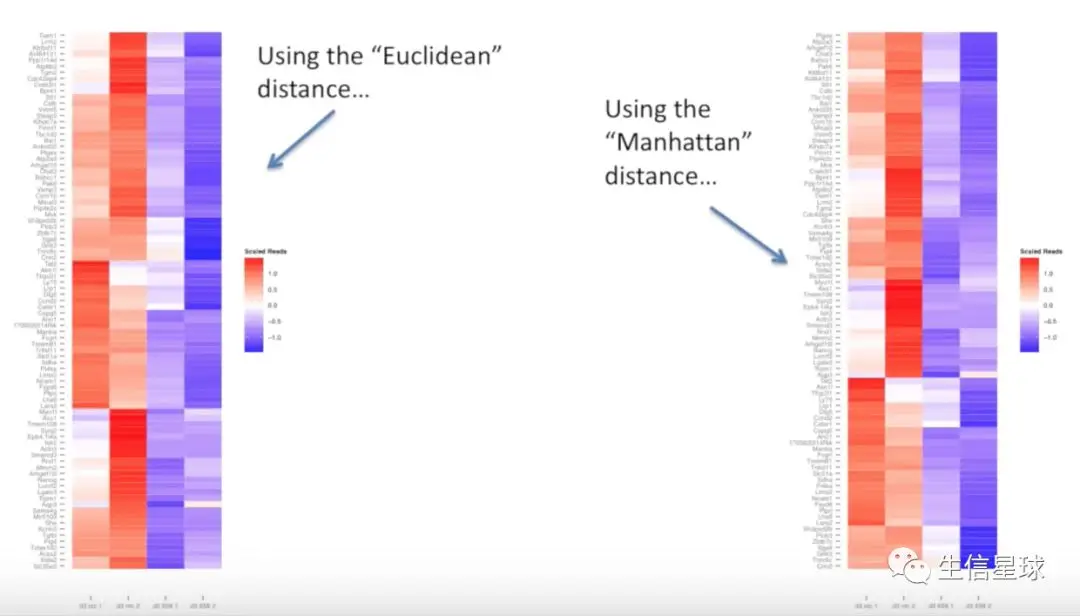
上面说的Euclidian method只是其中一种计算方式,还有其他的方法
比如 Manhattan distance:仅仅计算了差异的绝对值的加和,没有根号,没有开方
比较cluster
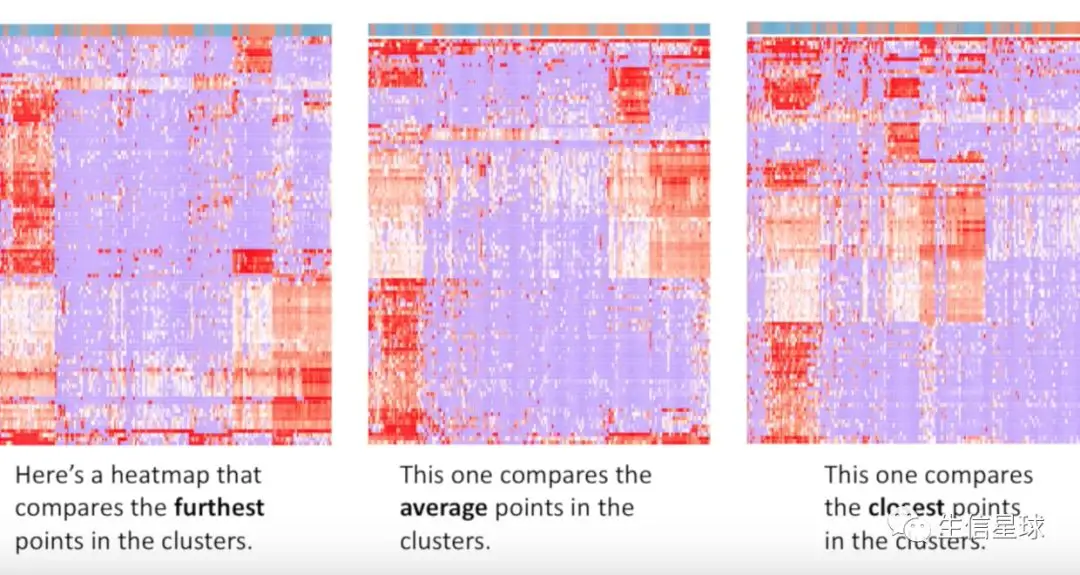
一般有三种:centroid(计算每个cluster均值), single-linkage(计算cluster最近的点), complete-linkage(计算每个cluster中最远的点)
R的hclust就是计算的最远的点