040-画个圈圈祝福你
刘小泽写于18.10.3 学习障碍的规律往往是:不是因为它多难,而是因为你不知道它难不难!
之前就见过各种文章中的复杂美观的圆圈图,被震撼到,这么多的信息是如何放到一个图中的?后来遇到一个网站Circos专门做圆圈图,很专业的样子,于是有心学一下。虽然看似很难,但是美好的东西,不都是奋斗出来的吗?
前言
背景知识
Circos并不直接用R做,而是一个Perl语言开发的可视化软件,由加拿大生信科学家Martin Krzywinski开发,可以用于描述关系型数据和多维度的数据可视化
其次,什么样的可视化软件会受欢迎?一是输入简单,不需要太多的数据处理技巧就能调整到要求的输入格式;二是输出美观、快捷。这两点,Circos都做到了
Circos的设计初衷:展示线性位置关系(如染色体上的信息)、展示一个几何中两个对象的关系(如比对信息)。将线性折成封闭曲线可以减少数据分散性

重造了个轮子?
程序设计者最大的忌讳就是“重复造轮子”,目前有许多基因组浏览器:UCSC(http://genome-asia.ucsc.edu/cgi-bin/hgGateway?redirect=manual&source=genome.ucsc.edu)、Ensembl(https://asia.ensembl.org/index.html)、Vista(http://pipeline.lbl.gov/cgi-bin/gateway2?bg=cin1&selector=vista)、VEGA(http://vega.archive.ensembl.org/index.html)、ARGO()。 当然,这些基因组浏览器含有的数据量都非常大,但如何从隐晦的庞大数据中抽出具体的数据再展示出来是个问题。比如染色体包含的数据很多,我们要是想展示染色体上的位置特征如某些基因,用circos的圆圈图就可以兼顾大数据与小信息
数据的关系
先说下数据类型,无非就是标量数据(scalor,有大小,没方向)、向量数据(vector,有大小和方向),将数据可视化也就是标量化、向量化。基因组就是一个一维的数据结构,上面的位置就是标量

最常见的就是标量数据进行标量可视化,比如输入数据是基因组位置,输出是实值(如GC含量、保守度),就可以用直方图(histogram)、折线图、散点图(scatter)画出。当数据集比较小、关系比较简单时还是可以看出变化趋势的,但是当数据之间存在交叉、数据量比较大时,一致性就体现不出来啦

或者有的图很好看,但是信息杂乱无章

又或者想表达相关性,一般使用曲线,但是在标量数据上展示曲线,数量一多就乱了

可能会想,把部分曲线抬高不就能区分开啦?但是抬高了显示的信息量势必会减少

为什么用圆圈?
图形可视化这么多形状,矩形、三角形、圆形…,其中圆形比矩形更对称,视觉压力不会那么大,看圆形会更圆润;
圆形有效载荷(data payload)会更大:文章中在尺寸、位置等对图片的限制还是蛮多的,如何体现数据有效载荷,这里有一个公式~轴的长度/图片面积。
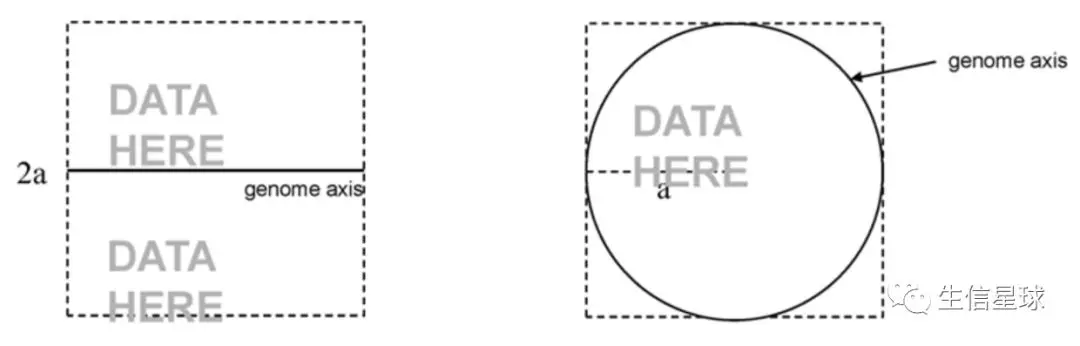
对于矩形来说,2a/4a^2^ = 1/2a (2a是x、y轴长度之和); 对于圆形来说,2πa/πa^2^ = 2/a。圆形是矩形体现的数据的4倍
轴长度多长会分散注意力,圆形更容易聚焦
看下面图是不是更好点?

圆圈图的一些元素
可以设置环形的数据轨道:如线形、散点、直方图,可以设置点(线)颜色、粗细、线形状。并且各个区域可以直接缩放,而不用截掉数据
环形坐标轴 需要带有距离刻度的对象,比如:染色体、序列、基因图谱、片段重叠群(contig)

配置文件,在内置的基础上进行修改,比如thickness、size、label、labelsize、grid等
使用高亮highlight创建不同的颜色片段,置于所有图层底部

基因组间的mapping 设置links seqdup,也就是不同染色体中出现的同一个序列片段,在圆圈图中进行展示,例如

设置格式规则

组合不同的图片类型 左:散点+箱线+折线;右:瓦片+热图+直方图

实际应用 输入:GFF;输出:PNG、SVG
安装
基于Unix(推荐linux)和perl,需要熟悉命令行
先检查Perl
Unix是默认安装了perl,perl 5.8之前的都需要升级;windows用户可以用dos或者git bash,先调出来命令行,然后安装perl,安装 Strawberry Perl 或者 ActiveState Perl perl在linux下的存在目录是/bin/env,在mac下的存在目录是/bin/usr/env
下载
# 目前最新的版本还是17年7月31的
wget http://circos.ca/distribution/circos-0.69-6.tgz
tar zxvf circos-0.69-6.tgz
# 放入环境变量
echo 'export PATH=/Your_path/circos-0.69-6/bin:$PATH' >>~/.bashrc
source ~/.bashrc
初步配置
(服务器可能需要root权限,用mac或者windows的命令行也是可以的)
# 配置cpanm,并加入环境变量
wget https://raw.githubusercontent.com/miyagawa/cpanminus/master/cpanm
# 先看看需要用到的perl模块,有一些缺失(提示:missing)的
circos -module
# 安装缺失模块
circos -module | grep 'missing' | awk '{a=a" "$2;}END{system("cpanm"a);}'
# 绕过root权限安装perl 模块
https://www.maketecheasier.com/install-perl-module-in-linux-without-root-permission/
perl模块缺失怎么办?
例如缺少Clone模块: 先在metacran网站查找:https://metacpan.org/pod/Clone 下载解压
wget https://cpan.metacpan.org/authors/id/G/GA/GARU/Clone-0.39.tar.gz
tar zxvf Clone-0.39.tar.gz
cd Clone-0.39
# configure
perl Makefile.PL
#Checking if your kit is complete...
#Looks good
#Generating a Unix-style Makefile
#Writing Makefile for Clone
#Writing MYMETA.yml and MYMETA.json
make
make test
make install
# 如果有使用build的模块,还是下载安装,然后
perl Build.PL
./Build
./Build test
./Build install
# 又或者使用conda安装
conda install -c bioconda perl-clone
安装的目录

bin:包含circos的可执行程序(已加入环境变量)
data:一个文件夹karyotype,包含几个物种的染色体信息,绘图必须

#chr - ID LABEL START END COLOR
chr - hs1 1 0 247249719 chr1
chr - hs2 2 0 242951149 chr2
chr - hs3 3 0 199501827 chr3
chr - hs4 4 0 191273063 chr4
chr - hs5 5 0 180857866 chr5
chr - hs6 6 0 170899992 chr6
#...省略部分
band hs1 p36.33 p36.33 0 2300000 gneg
band hs1 p36.32 p36.32 2300000 5300000 gpos25
band hs1 p36.31 p36.31 5300000 7100000 gneg
band hs1 p36.23 p36.23 7100000 9200000 gpos25
# ...
#karyotype.human.hg18.txt
前两列:chr定义染色体,-表示父区域,染色体没有父区域,因此用-,下面的band就有父区域了,比如这四行的band的父区域就是hs1,1号染色体;
ID是命令,同时绘制多个物种,可在ID中包含物种的代号;
LABEL:当前区域显示的名字;
START END:整个的区域范围,要显示部分区域,可以后来修改;
COLOR:当前区域的颜色,定义好的颜色,存储于etc/colors.conf
etc:配置文件,如
housekeeping.conf #基本框架配置 fonts.conf #字体配置文件 background.black.conf #只定义了背景是深色 background.white.conf colors.hsv.conf colors.ucsc.conf colors.conf #利用RGB组合设置了颜色变量、系列颜色和染色体的颜色 colors_fonts_patterns.conf #颜色、字体、预定义的图形文件 image.conf #设置image大小、背景颜色、输出目录、输出文件形式以及染色体在圆周上的起始位置等的设置 ideogram.conf #显示每个染色体的配置文件,包括是否展示bands、染色体位置展示、染色体间距离、染色体标签位置等(bands指的是在染色体组上加上条纹图谱信息,染色体图谱看上去有被染色的效果) image.generic.conf patterns.conf #定义特殊小图形的信息 ticks.conf #刻度标签配置文件,以刻度形式显示染色体的大小 links.conf #以曲线连接显示基因组内部区域间的联系 histogram.conf #以直方图展示数据 heatmap.conf #以热图展示数据 ...
刘小泽写于18.10.4
自己安装最大的问题就是:perl模块配置,不管自己的机器还是服务器,总是有模块没有配置好,需要一步步调试;而且模块相互之间需要依赖,又是个麻烦事。
要图方便,使用conda安装circos也是可以的,省去不少麻烦,三步搞定
conda create -n circos source activate circos conda install circos -y # 最后用circos -modules检查一下 # conda安装的目录在 miniconda3/pkgs/circos-0.69.6-2
使用
官方教程 http://circos.ca/documentation/tutorials/
使用过程并非可视化的,需要以下几步: 想想数据要被怎么展示(最麻烦的部分); 将数据转换成circos要求的格式; 构建配置文件,可以用默认的当作模版进行修改; 运行circos生成PNG或者SVG; 微调PNG/SVG,例如添加图例、文本标题等
配置文件概况
circos生成的是静态文件,这个过程是配置文件起主导作用,例如全局的字体、颜色设置,这个过程需要Config::General这个模块。其中的配置信息用空格作为分隔符,使用=定义变量名 = 变量值。有的一个相同的配置需要分成几个小配置块,是为了给不同的track(圆圈轨道)个性化定制,比如
<links>
# track 1
<link>
file = data/set1.txt
color = black
...
</link>
# track 2
<link>
file = data/set2.txt
color = red
...
</link>
</links>
开始符<>与结束符</>组合成一个配置块(二者缺一不可),几个重要的配置块放在一个配置文件中
<ideogram> # 例如开始是这个板块
...
</ideogram> # <-- 结尾没有这个就报错
配置文件中有4个地方可以自定义:(排序:优先级越来越高)
- 全局实例:所有图形中的所有数据点(位于
<plots>或者<links>配置块) - 局部实例:某个图形中所有数据点(位于
<plot>或者<link>配置块) - 数据:指定某个数据点(位于
data文件) - 规则:位于
<rule>配置块【比如这里的设置可以覆盖之前的数据的设置】
一个全局实例可以包括多个局部实例,比如下面配置文件中fill_color = gray对全局和局部都有效,而fill_color = white 只对第一个局部实例内部有效。当许多局部实例组合在一起时,使用全局实例能提高效率
<plots> #全局plot
fill_color = gray
...
<plot> #plot1,比如这个可以负责画环
fill_color = white
...
</plot>
<plot> #plot2,比如这个可以画点
...
</plot>
</plots>
利用内置的配置文件
位于circos/etc的目录下,这些配置文件都是优化好的,基本不用修改直接写入自己的配置文件。
常用的几个不需要更改的:
housekeeping.conf基本的框架配置,直接导入fonts.conf配置字体,与font目录下的ttf或otf文件对应pattern.conf配置输出格式,与tiles目录下的png文件对应,然后用pattern.svg.conf生成svg文件colors.conf:引入了colors.brewer.conf、colors.hsv.conf、colors.ucsc.conf定义了颜色的不同绝对路径,另外在本身的colors配置文件中,还支持使用像red、hs1、reds-9-seq-1的方式
几个重要的需要稍微调整的:
image.conf调整图片大小、背景颜色、输出目录、输出文件格式、染色体在圆圈的起始位置ideogram.conf绘制基因组的核型数据——染色体,例如人,会把22条常染色体和X、Y绘制出来,而且在最外圈展示,每条染色体都是一段圆环。专门对染色体进行配置:染色体间距、标签位置、是否展示bandsticks.conf刻度形式显示染色体大小,对刻度进行配置,可以导入ideogram.conf
配置文件导入:<<include xxx.conf>>省去了主文件重复定义,可以直接引用其他的配置文件,层层引用嵌套,例如下面的配置文件逻辑关系就比较清楚:
karyotype = data/karyotype/karyotype.human.txt #指定物种是人
chromosomes_units = 10000000
chromosomes_display_default = yes
<<include ideogram.conf>> #引用:链接核心图——染色体作为外圈
<<include ticks.conf>> #引用:加刻度
...
<image> #输出图片
<<include etc/image.conf>> #引用:图片设置
</image>
<<include etc/colors_fonts_patterns.conf>> #引用:设置颜色
<<include etc/housekeeping.conf>>
画出第一个轮廓图/核型图(没有数据,只有染色体的idogram)

1. 先准备好被导入的配置文件:circos/etc目录下文件可以直接用

2. 再创建ideogram.conf和ticks.conf配置文件
# ideogram.conf
<ideogram>
<spacing>
default = 0.005r # 1.设置两条染色体间空隙,每个空隙大小为周长的 0.5%(这里的r表示ring圆环,而不是半径)
</spacing>
radius = 0.9r # 2.1 染色体所在位置(即画在多大的半径上)在离圆心的 90% 处 (这里的r指的是半径,而不是ring)
thickness = 20p # 2.2 染色体厚度
fill = yes
fill_color = black # 2.3 染色体颜色
stroke_thickness = 2 #2.4 染色体边框厚度
strock-color = black
show_label = yes # 3.显示染色体标号如CHR1
label_font = default
label_radius =1r + 75p # 1r对应ideogram对应的半径,+75p表示在圆圈外侧75p(像素pixel)的地方;如果要显示在内侧,可以1r - 75p或者0.9r类似这种
label_size = 30
label_parallel = yes #设定 label 的字体方向,yes 生成易于浏览的方向
label_case = upper #label大小写显示
show_bands = yes # 4.染色体上条带bands是否显示
fill_bands = yes
band_stroke_thickness = 2 # 边框厚度
band_stroke_color = white #颜色
band_transparency = 4 #透明度
</ideogram>
关于单位设置:共有4种单位:p, r, u, b。p表示像素,1p表示1像素;r表示相对大小,0.005r表示0.5% ring 大小;u表示相对chromosomes_unit的长度,如果chromosomes_unit = 1000,则1u就是千分之一的染色体长度;b表示碱基,如果染色体长1M,那么1b就是百万分之一的长度
# ticks.conf 刻度形式显示染色体大小
show_ticks = yes # 1.是否显示刻度
show_tick_labels = yes
<ticks>
skip_first_label = no # 2.刻度所在位置、颜色、厚度
skip_last_label = no
radius = dims(ideogram,radius_outer)
tick_separation = 3p
label_separation = 1p
multiplier = 1e-6
color = black
thickness = 4p
size = 20p
<tick>
spacing = 1u # 3.次刻度
show_label = no #不显示次刻度标签
thickness = 2p
color = dgrey
</tick>
<tick>
spacing = 10u # 4.主刻度
show_label = yes # 显示主刻度标签
label_size = 20p
label_offset = 10p
format = %d
grid = yes
grid_color = dgrey
grid_thickness = 1p
grid_start = 0.5r
grid_end = 0.999r
</tick>
</ticks>
3. 创建主配置文件,并导入之前设置的其他配置文件
保存为任意名称(这里以circos.conf为例)可以直接复制粘贴后,用circos -conf circos.conf得到circos.png、circos.svg文件
# 首先karyotype核型文件是必须的,定义了需要染色体的名称、大小、颜色,并且可以根据自己的需要设定。不限于染色体,还可以是contigs、genes等可以在坐标上体现出来的。circos自带的核型文件放在/data/karyotype下
karyotype = data/karyotype/karyotype.human.txt
chromosomes_units = 10000000 #指定距离单位u
# chromosomes = hs1;hs2;hs3这样写表示只显示三条染色体(因此下面的👇设置要写成no)
chromosomes_display_default = yes #显示所有的染色体(no的话需要自己指定)
<<include ideogram.conf>>
<<include ticks.conf>>
<image>
# file = xxx 给图片命名
# dir = /tmp
# 24bit = yes
# png = yes
# svg = yes
<<include etc/image.conf>>
</image>
<<include etc/colors_fonts_patterns.conf>>
<<include etc/housekeeping.conf>>
核型图内侧添加数据
数据图包括许多种:散点图、线形图、柱状图、热图,R能画的图基本Circos都支持;另外还可以画联系图Connectors、高亮图Highlights、连接图Links(比如某些融合基因存在于1区域和2区域,就可以连接这两个)
散点图、线形图、柱状图、热图数据格式
# chr start end value option(可有可无)
...
hs1 0 66666 0.01314 #其中0-66666相当于x坐标,0.01314相当于y坐标
hs1 66667 166666 0.00520
hs1 166667 266666 0.00231 fill_color=red
#fill_color=red,z=66就是option部分,可有可无,表示设置这一块区域显示什么颜色等等,还有其他一些设置
...
散点图
写好散点图的配置文件,加到主配置文件中即可
<plots>
type = scatter #在全局设置可以应用一下好几个plot,就省去了一个个设置的麻烦。首先设置绘图类型,可以是scatter、line、histogram、heatmap
stroke_thickness = 1
<plot>
file = data/6/snp.density.txt #指定数据文件
fill_color = grey
stroke_color = black
glyph = circle #点形状及大小
glyph_size = 10
max = 0.013 #设置数据范围,超出的不考虑
min = 0
r1 = 0.95r #绘制区域,r0是内半径,r1是外半径。plot1是从圆圈内部画图
r0 = 0.65r
<backgrounds> #设置plot1背景颜色(点密集区域一个颜色,此处用灰色;点稀疏区域用绿色区分)
<background>
color = vvlgreen #颜色设置:very very light green (vvlgreen)
y0 = 0.006 #y0设置背景色起始位点,可以使用绝对值比如这里的0.006,也可以使用相对值,如0.75r
</background>
<background>
color = vlgrey
y1 = 0.006 # y1设置背景色终止位点
y0 = 0.002
</background>
</backgrounds>
</plot>
<plot>
file = data/6/snp.density.txt
#需要的数据都在https://github.com/vigsterkr/circos/tree/master/data/6
# 或者git clone https://github.com/vigsterkr/circos.git
fill_color = green
stroke_color = dgreen
glyph = rectangle
glyph_size = 8
max = 0.013
min = 0.007
r1 = 1.175r #plot2是从圆圈外部画图;绘图区域方面,不同的plot一般不会设置重叠
r0 = 1.075r
</plot>
</plots>

线形图
线形图和散点图类似,只不过线形图用线代表了点,纵坐标大的点在线形图中就是一条细高的线
<plots>
type=line
thickness = 2
<plot> # plot1
max_gap = 1u
file = data/6/snp.density.250kb.txt
color = vdgrey
min = 0
max = 0.015
r0 = 0.5r
r1 = 0.8r
fill_color = vdgrey_a3
<backgrounds> # plot1背景色
<background> #
color = vvlgreen
y0 = 0.006 #y0设定最低阈值,将高于0.006的部分设成淡绿色
</background>
<background>
color = vvlred
y1 = 0.002 #y1设定最高阈值,将低于0.002的部分设成淡红色
</background>
</backgrounds>
<axes> #设置坐标轴,这个轴是沿着圆圈一圈一圈绕的,就像老唱片的磁道一样
<axis>
color = lgrey_a2
thickness = 1
spacing = 0.025r #设置坐标轴间距
</axis>
</axes>
<rules>
<rule>
condition = var(value) > 0.006 #画出高于0.006的值,这部分值显示在绿色区域中
color = dgreen
fill_color = dgreen_a1
</rule>
<rule>
condition = var(value) < 0.002 #画出低于0.002的值,并且这些值显示在红色区域中
color = dred
fill_color = dred_a1
</rule>
</rules>
</plot>
<plot> # plot2
max_gap = 1u
file = data/6/snp.density.txt
color = black
min = 0
max = 0.015
r0 = 1.075r
r1 = 1.15r
thickness = 1
fill_color = black_a4
</plot>
</plots>

直方图
它是线形图的变体,线形图中就是用直线把近似的点连接,直方图中就是把点用一个个方块表示
<plots>
<plot>
type = histogram
file = data/6/hist.random.txt
color = red
r1 = 0.9r
r0 = 0.7r
max = 1 #这个看数据具体范围设定
min = -1
thickness = 10p
fill_under = yes
fill_color = red
orientation = out #控制方向(红色向外)
</plot>
<plot>
<plot>
type = histogram
file = data/6/hist.random.txt
color = green
r1 = 0.6r
r0 = 0.4r
max = 1
min = -1
thickness = 10p
fill_under = yes
fill_color = green
orientation = in #同时展示下向内的直方图(这个功能就是为了区别不同的数据集)
</plot>
</plots>
使用的测试数据只有3条染色体信息,所以先画三条的,明白怎么画最重要

热图
要求的数据依然是:
# chr, start, end, value, and a list of optional parameters
...
hs7 36975000 36999999 33
hs7 37000000 37024999 50
hs7 37025000 37049999 60 color=blue
hs7 37050000 37074999 44
...
热图最大的特色就是利用颜色来区分数据,因此设置了color这个选项;并且颜色都是可以多种多样的,具体的可以参考官方关于颜色的说明
<plots>
type = heatmap
<plot>
file = data/6/snp.number.1mb.txt
color = spectral-9-div #产生11种颜色,并且根据从min到max的数据赋予不同颜色。这个色板的信息位于etc/brewer.conf
stroke_thickness = 1
stroke_color = black
min = 1000
max = 5000
r0 = 0.80r
r1 = 0.90r
</plot>
<plot>
file = data/6/variation.heatmap.txt
color = spectral-9-div
stroke_thickness = 1
stroke_color = black
min = 2000 #为了能使颜色区分开,这里的min和max的设置非常重要,如果设置的间距太小,就容易区分不明显
max = 250000
r0 = 0.60r
r1 = 0.70r
</plot>
<plot>
file = data/6/variation.heatmap.txt
color = ylorrd-9-seq-rev
stroke_thickness = 1
stroke_color = black
min = 2000
max = 250000
r0 = 0.40r
r1 = 0.50r
</plot>
</plots>

联系图Connector
这种图会把核型图上位于同一个染色体的起始和终止位点连接,不能跨染色体
输入数据格式是:染色体 起始 终止
另外有一个特定的参数:connector_dims 需要设定5个值,分别代表画线起始点、直线、斜线、直线、画线终止的比例,并且5个值的和是1
<plots>
<plot>
type = connector
file = data/6/connectors.txt #要注意不同的数据使用的染色体不同,适时调整ideogram中的染色体配置,比如是否要显示全部染色体,要展示哪几条染色体等
r0 = 0.70r
r1 = 0.90r
connector_dims = 0,0.3,0.4,0.3,0 #表示开始画图=》第一条直线比例是0.3=》斜线比例是0.4=》第二条直线比例是0.3=〉画图终止
thickness = 2
color = red
</plot>
<plot>
type = connector
file = data/6/connectors.txt
r0 = 0.40r
r1 = 0.60r
connector_dims = 0,0,0.7,0.3,0 #斜线占的比例越大,倾斜角度越小
thickness = 2
color = black
</plot>
</plots>

高亮图Highlights
想强调某一个条带的某一个位置
数据格式是:染色体 起始 终止 颜色(fill_color = colr)
可以用<plot> + <type = highlights>的方式,也可以用<highlights>的方式
连接图Links
比如想看一个基因在一个染色体与另一个染色体上发生融合的现象,用弧线表示
数据格式:1stChr start end 2nd Chr start end 会将第一个染色体的终点和第二个染色体的起点连接起来
<links>
<link>
file = data/5/segdup.bundle3.colored.txt
color = red
radius = 0.95r # 设定弧线起始点的位置位于0.95r的位置
bezier_radius = 0.1r # 控制弧线弧度
thickness = 1
</link>
file = data/5/curves.repeated.txt
color = blue
radius = 0.95r
bezier_radius = 0.1r
thickness = 1
</links>

连接图升级版—丝带图Ribbon

终极组合图
代码在http://circos.ca/documentation/tutorials/2d_tracks/stacking_tracks/configuration。当然需要细细品味其中的意义,但是类型就是这几种
