034-关于基因的富集分析
刘小泽写于18.9.14,更新于18.9.22
不管是转录组,还是芯片数据,或者其他有关基因的组学分析,每当数据分析到后面,要想得到结果,都躲不过这个富集分析,因为它是帮助我们从庞杂的组学数据中发掘规律重要的一环。对基因功能进行富集分析, 就有可能发现在生物学过程中起关键作用的生物通路, 并且帮助理解生物学过程的分子机制
现在的高通量测序带来的巨大数据量,让我们眼界大开,局限于单纯的某个基因的做法越来越行不通,但是想要从庞大的关系网络中挑选出有效信息,比如将某几个基因和某个期待的生物学现象结合起来,这个事直接做是很困难的。因此为了降低研究的复杂度,将不同生物学现象与基因的对应关系做成了多个数据库。于是,当我们手上有成百个差异基因时,就去不同数据库比对,这个过程就叫做富集分析。
简而言之,基因富集分析 是在一组基因中找到具有一定基因功能特征和生物过程的基因集,在研究差异表达基因、筛选基因的后续分析中经常使用。
基因集,也叫gene set,也就是一系列具有相同功能的基因构成的集合,比如某一条代谢通路(pathway),其中有很多的基因,因此位于同一条通路下的基因就构成了一个基因集合。 组成基因集的最基本元素就是一个一个的基因,在芯片分析中,结果往往是差异表达的探针,需要先将探针映射到基因上。注意:在映射的过程中,必须考虑到基因和探针之间的对应关系,会有多个探针对应一个基因的情况,虽然比重不大,却还是要考虑。比如分析甲基化数据时,由于大部分的基因具有多个CpG位点,因此会对应多个探针ID。有时A、B基因都有探针比对上,但不能就这样认为他们的差异量一样,因为A、B的差异CpG位点有时不同,A有30个差异CpG位点,B却只有3个,他们虽然都叫差异基因,但差异也分大小,不能一概而论
富集分析目的
万事万物皆有其因,事实上,我们做这个富集分析的目的主要包括:
目前正在研究某个基因,想看它在不同样本中的表达差异,也就是我们有和课题相关的目标基因,你认为这个基因是处理和对照产生不同的原因之一,但是口说无凭,需要佐证。此时你需要富集分析
跑程序得到的差异基因上千个,你想知道他们是那些类的,和物种什么生物过程相关,你会把基因一个一个放到注释数据库去调查吗?此时你需要富集分析
研究某个基因的上下游调控关系时,你可能对KEGG的那个通路图不陌生,这就是富集分析
它是快速调查目标基因集功能倾向性的方法之一。因此你可能还听过通路分析(pathway)、功能分析。就是说,手里有基因、蛋白的,都要经历这一步
举个生活中的例子:城市中都有生活公园,清晨你会看到许多老人在锻炼身体,上午游人来玩耍,小商贩也不能放过这个机会,下午到了放学的点,孩子们蜂拥而至,而到了晚上,中年人群广场舞激情飘扬。现在想看看公园哪个时间段更吸引人。假如一天来公园的共1000人,现在把这些人都汇集在一起,从中抽取200人,结果看到70%以上都是老人,那么基本可以确定清晨公园人流量更大。然后针对主要公园人群——老年人,公园管理部门就可以安排更便民的设施。当然,这个例子只是为了理解下面的内容
我们上面不同年龄的人群,就对应不同功能的基因集,当然人群中的每个人都可能不同时间光顾公园,当然基因集中的不同基因也可以参与好几个生物过程。我们这里做的富集分析,不是为了分析个体,而是看群体。先判断出哪些群体的差异是我们想要的,再看其中的个体~就是这么个过程!
富集分析算法
任何软件、分析背后都是一套算法,了解算法才能真正坐观云卷云舒
具体的方法介绍可以参考:Progress in Gene Functional Enrichment Analysis这篇文章。大体上富集分析有四类算法:ORA、FCS、PT、NT
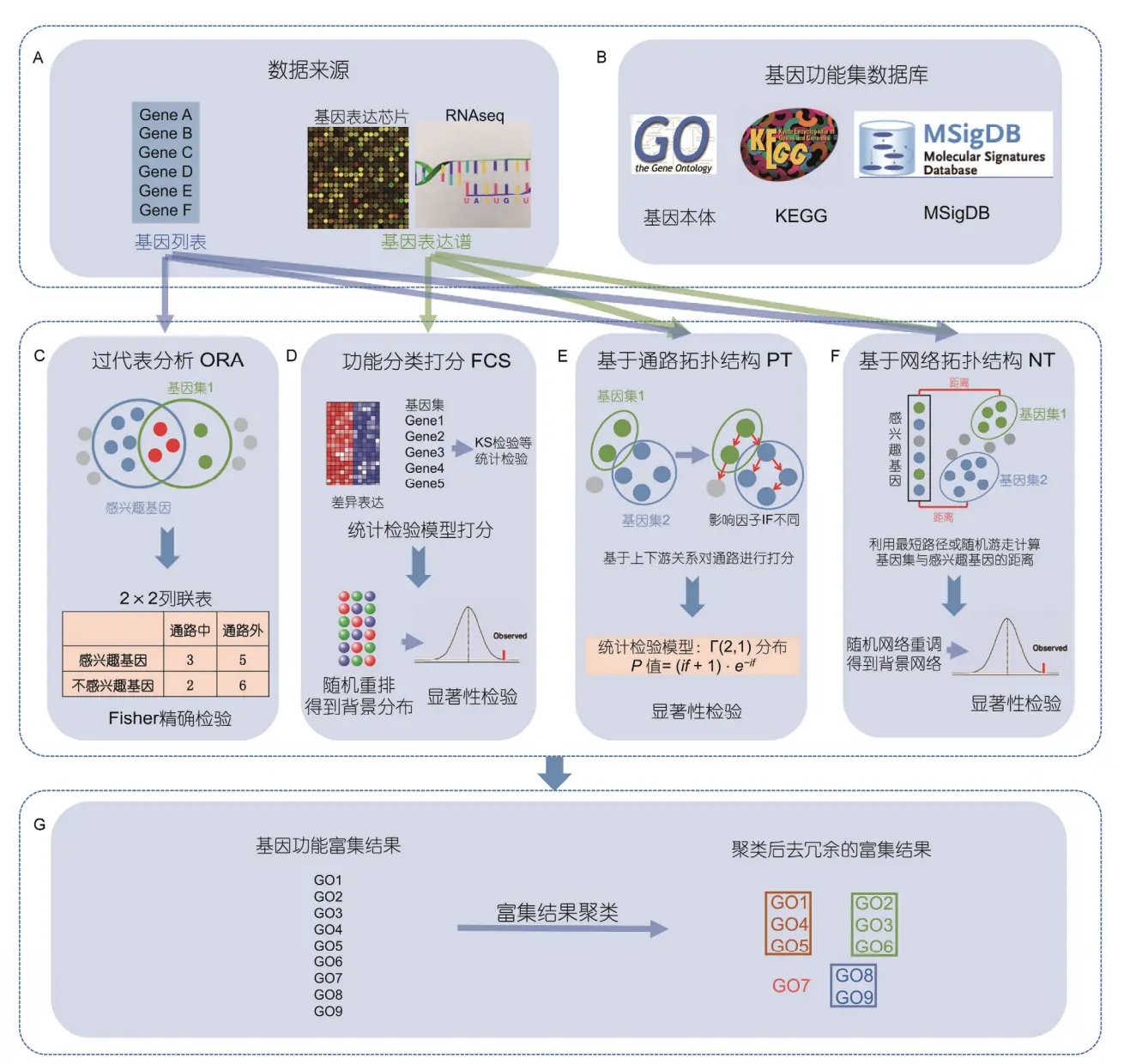
1. ORA(Over Representation Analysis):过表达分析
首先这个名字很奇怪,但是既然人家取了这个名字,就一定有原因。
这个部分很重要,需要重点理解,采用“理论联系实际”的方法
理论:
什么是ORA方法? 它是检验某类功能在一个数据子集中是否表现过度。又称为“2X2方法”,像上图一样,做一个列联表。上图中的ORA中,蓝圈内是感兴趣基因(8个),绿圈内是某个通路的基因(5个);灰点是既不感兴趣又不在通路内的(6个),蓝点是感兴趣但不在通路内的(5个),绿点是在通路内但不感兴趣的(2个),红点是既感兴趣又在通路内的(3个),于是就能做出来2X2列联表。再利用fisher精确检验或超几何分布得到p值。
好啦,看到概率分布,先去补习背景知识~
fisher精确检验是基于超几何分布计算的,包括:单边检验(类似于超几何检验)和双边检验; 二项式分布,就是抽取n个东西,其中k个是符合要求的,但它是有放回抽样,想象一下,你能从成千上万基因中多次抽到同一个基因,这是不符合常理的;
简而言之,需要4类数据:总共的基因数(作为背景基因)、总共属于某分类的基因数、样本包含的基因数(也就是用的差异表达基因)、样本中属于某分类的基因数
优点:出现的最早,最常用,有完善的统计学理论基础,结果比较可靠;
缺点:
- 仅仅使用了基因的数目,但是基因的不同表达水平没有考虑,为了得到差异基因,需要人为设置阈值,没有一个设置规定,因此结果因人而异;
- 适用于差异最显著的基因,而差异不显著的基因就会被忽略,检测灵敏度会降低
- ORA利用统计学假设每个基因相互独立,但是就生物体本身而言,忽略了内部的复杂的相互作用,并且每个基因在不同的生物学过程中发挥的作用大小不一样,同等看待结果可能会不准确
实际:
实际上就是把我们感兴趣的基因和背景基因做一个交集。
感兴趣的基因也就是差异基因了,包括上调、下调表达的(利用原始表达矩阵中p值和logFC进行筛选),一般人类芯片数据会有几百个
背景基因就是在KEGG等数据库中有注释的基因【人类基因组有2万个左右基因,现在总共有已知功能的是7000左右,随着研究的不断深入,背景基因数量会越来越多,结果也会越来越全面】

举个例子,KEGG通路hsa05206指的是MicroRNAs in Cancer,包括150个基因,背景基因使用了6517个;GSE17708芯片得到的差异基因数是547个,在KEGG能注释上的有80个,其中就有10个是MicroRNA通路的,概率高达12.5%(enrichKEGG方法都是用能在KEGG注释上的基因,比如这里用80而不是547),那么这个通路是不是在下调基因中被显著改变?需要把全部的80个下调基因,在KEGG的530个通路中注释一遍,再一个一个进行超几何分布检验,得到p值。hsa05206通路在背景基因中查到的概率是150/6517=2.3%,是显著低于12.5%的
超几何分布属于统计学上一种离散概率分布。它描述了由有限个物件中抽出n个物件,成功抽出指定种类的物件的个数(不归还)。n=1,超几何分布还原为伯努利分布;n接近∞,超几何分布可视为二项分布
2. FCS(Functional Class Scoring) 功能集打分法
它比ORA的进步就是基本假设做了改变,考虑的更加全面。它认为尽管单个基因的改变会造成显著性影响,但是和它类似的微效基因叠加在一起也能行。也就是说,FCS不再像ORA一样,强调个人英雄主义,而是把目光转向人民,“星星之火,可以燎原”
Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges这篇文章有详细的描述。
操作方法:
要求的输入是一个排序的基因列表和一个基因集合,不需要设置阈值
- 计算单个基因表达水平的统计值,采用如衡量差异基因的ANOVA、Q-statistic、t检验、Z-score、信噪比,进行打分或排序,或者直接使用排序好的基因表达谱
- 同一通路上所有基因的表达水平统计值进行整合,汇集成单个通路水平的分数或统计值,采用基因水平统计的和、均值或中位数,Wilcoxon rank sum, Maxmean statistic, Kolmogorov-Smirnov statistic
- 对通路水平的显著性进行评估:利用重抽样(bootstrap)的统计学方法
优点:
考虑了基因表达值的个体差异化信息,更加全面
缺点:
FCS仍然和ORA一样,只能独立分析每一条通路,但是同一个基因可能设计多个通路,不能分析这种情况;它只是根据特定的通路为差异基因进行排序,比如按基因A、B的一条通路检测得到他们的表达量分别改变3倍、10倍,但是换其他通路,可能排名就发生改变,不会一直保持B>A
3. PT(Pathway Topology)通路拓扑学
理想很丰满,现实很骨感的一个体系
在通路的富集分析中,一般上游基因的表达水平改变要显著大于下游基因对整个通路的影响。PT方法就是把基因在通路中的位置,和其他基因的互作和调控关系结合在一起,评估每个基因对通路的贡献并算出权重,然后把权重整合到富集分析。 方法虽好,但是通路拓扑结构存在依赖性,而目前的GO等数据库中没有任何拓扑结构信息,因此限制发展
4. NT(Network topology)网络拓扑学
利用现有的全基因组范围的生物网络,提取数据库的基因相互作用关系(如:基因连接度、基因在网络中的距离),把基因的生物学属性整合到功能分析。利用网络拓扑结构来计算基因对特定生物通路的重要性并给予相应的权重,再利用传统的ORA 或 FCS 方法来评估特定生物通路的富集程度,如GANPA 和 LEGO。缺点就是算法太复杂,计算速度慢
富集分析内容
一般提到富集分析,首先想到的就是GO、KEGG这两把刷子,然后还需要知道两个重要概念:前景基因、背景基因
前景基因:你关注的要重点研究的基因集;背景基因:所有的基因集 比如做转录组测序,一般都要设置处理组和对照组,前景基因是处理和对照的差异基因,背景基因就是两组样本的全部表达基因。
GO富集分析
Gene Ontology: 描述基因的层级关系【基于ORA算法】可以算得上是高通量数据分析的标配,转录组、甲基化、ChIP-seq、重测序等,都会用到对一个或多个集合的基因进行功能富集分析,来找这个基因集的功能偏好性
刚开始看到这两个词汇,我就看第一个单词比较熟,至于第二个嘛,“本体论”什么鬼?
本体论,既然是本体,那么就要求全,并且权威。因此,它就是某个科学领域中的专业词汇的集合,并且经过多方考证,能被公认的词汇表
GO发展 生物数据库有很多,但是不同的数据库之间可能会使用不同的术语,大量的研究时间都被浪费在查找信息上。于是基因本体联合会组织(Gene Ontology Consortium)建立了GO数据库,规范统一了对于不同物种的基因和蛋白描述。整个数据库前身就是1988年三大数据库的整合(果蝇数据库、酵母数据库、小鼠基因组数据库),现如今,已经支持几十种物种
发展30年,条目一定很多,有没有什么命名规律?
条目标准定义:
id:也就是GO编号,如:GO:0031985name:全称,Golgi cisternaontology:命名分类,cellular_componentdefinition:定义,Any of the thin, flattened membrane-bounded compartments that form the central portion of the Golgi complex. Source: GOC:mah除了这些标针对每个条目term的标准定义,还要定义条目之间的关系,让他们形成一个有结构的框架,而不是一盘散沙。于是GO采用了“**有向无环图”**的模式(有向指的是term之间的单向指向性关系,比如termA是内质网,termB是细胞器,规定A是B,却不能说B是A;无环指的是从任何一点开始沿着规定的指向都不能回到原点)表示这种关系的方式有三种
is_a/part_of/regulates【详见官网http://www.geneontology.org/page/ontology-relations】is_a和part_of都具有传递性,比如A is a term of B / A is part of B;regulate表示一个过程受另一个过程的影响,前者调控后者能简单点吗?主要包括啥?
它将基因分门别类放入细胞组分CC、分子功能MF和生物过程BP三个功能类别中(分别对应基因产物在哪里发挥功能、怎样发挥功能、发挥什么样的功能)
基因产物被尽可能的富集到最低层的功能term上。如果一个基因产物被定为在Golgi cisterna(高尔基池),那么它只会定位到这个特定的条目,而不会定位到它的父节点或其他上级条目中
GO的具体作用?
帮助我们找到提交的基因集中各个基因是否有共同的GO条目,或者有没有共同的上级GO条目,可以发现具有某些共同特点的基因(比如说是某类细胞器的组成成分,或在某个催化反应起作用,又或者在某条通路中起作用)
GO气泡图可以表示显著性不同的GO条目对应的基因数量;
GO条形图可以表示三个分类的前多少条目对应基因的数量,可以每个分类单独做,也可以组合在一起;
GO网络图可以表示显著性较强的GO条目之间的相互关系(点大小代表条目内基因数目多少,颜色代表P-value,值越小越红);
GOmap图可以表示显著性较强的GO条目的层级关系,以树状图的形式展现
GO数据库是不断更新的,因此GO富集结果和数据库使用关系密切,有时候公司给你返回来结果,你却找不到想要的基因,除了实验因素,有可能是注释环节采用的工具没有及时更新,采用了老版本的软件和数据库。比如Nature methods就在2016年发表了一篇Impact of outdated gene annotations on pathway enrichment analysis,吐槽DAVID在线工具的数据库老旧,更新慢的问题。再比如Y叔曾经吐槽过华大的WEGO数据库还是09年的。虽然做出来图是那么回事,但是解释不了当前的问题。其实数据库并没有错,GO注释每天都在更新,Pathway数据库例如Reactome和PathwayCommons每个季度都在更新。 因此,建议不要太依赖在线工具,因为它的维护不一定那么及时
KEGG
Kyoto Encyclopedia of Genes and Genomes: 系统分析基因产物和化合物在细胞中的代谢途径以及这些基因产物的功能的数据库【基于ORA算法】
它的产生就是因为大量的数据被获得,却埋没在浩如烟海的文章之中,没有人去归档整理,但是使用这些数据的人还不少,他们每次都在检索上花费了大量时间。而日本的商人看到了商机,他们的收纳功力世界文明,于是把很多数据分类整理,做成了一个付费的平台,这开始就是一个“卖方市场”,十分抢手。但是到如今,其实大量的开源软件对它构成了威胁
包括什么?整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等等。KEGG 有一套完整KO注释的系统,可完成新测序物种的基因组或转录组的功能注释(KO是蛋白质或酶的一个分类体系,将同一条通路上功能相似、序列相似的蛋白质归为一类)。因此它可以将基因一个个归置到代谢网络指定位置上。
命名规则:
K(大写) +num基因ID号,表示所有同源物种中具有相似结构或功能的一类同源蛋白,如:K04456表示丝氨酸蛋白激酶;ko+num代谢通路,表示特定的生物路径,如:ko04151表示PI3K-Akt信号通路【也是我们常用的代谢通路】;M+num表示模块,如:M00676表示PI3K-Akt信号模块C+num表示化合物,如:C00533表示一氧化碳ECx.x.x.x表示酶,如:EC2.7.11.1表示丝氨酸R+num表示反应名称关于物种缩写:https://www.genome.jp/kegg/catalog/org_list.html
举个例子:三个字母表示物种,
hsa表示Homo sapiens;具体的KO号,如K12407表示和葡糖激酶glucokinase序列和功能相近的蛋白质/酶类,当然一个KO号有可能会对应好几个数字(基因登录号),表示细胞中存在几种不同的葡糖激酶,分别由以上几种数字表示的基因编码怎么使用该图?
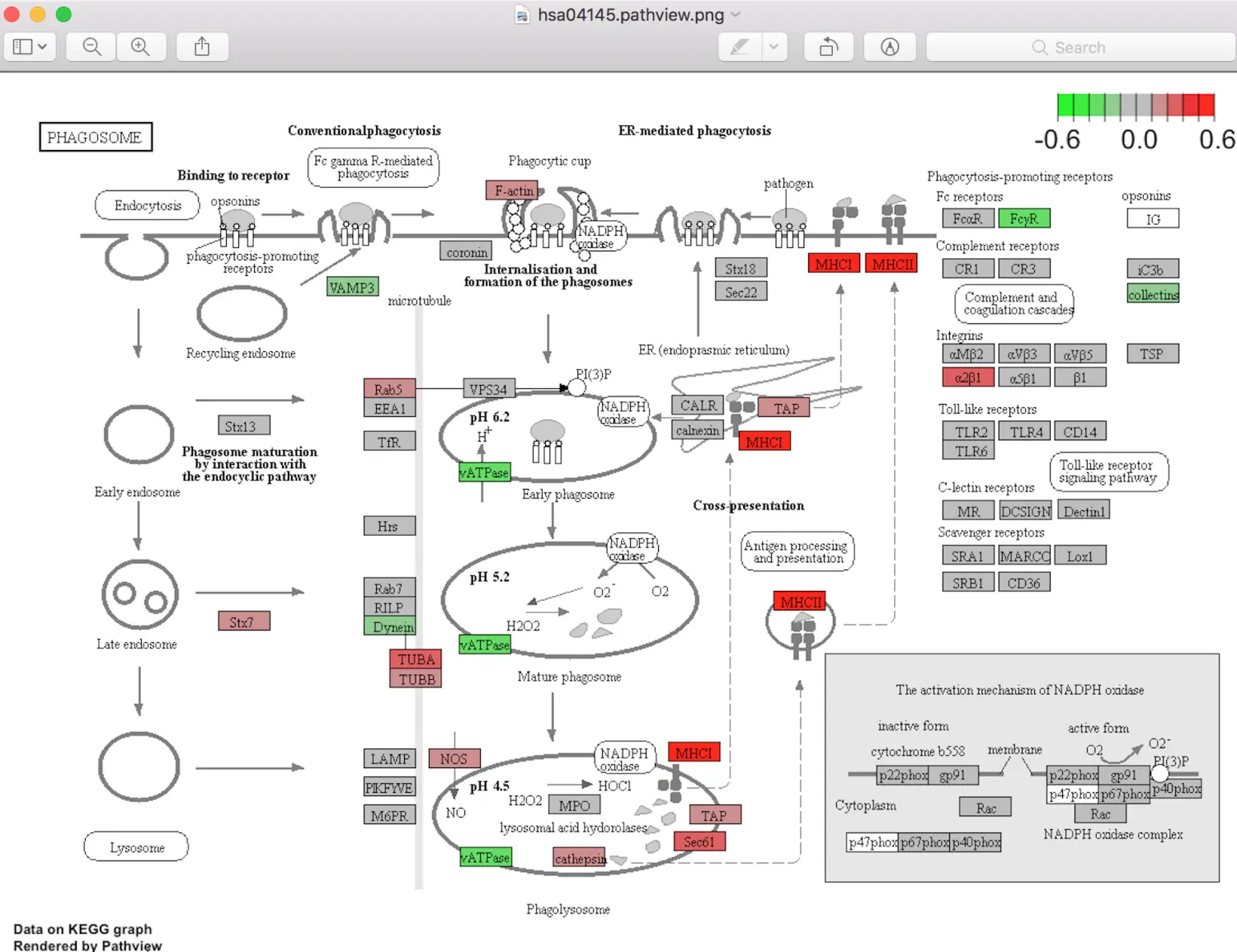
这个图可以用pathview函数获得。先看看有没有关注的基因能注释到通路上,主要看颜色:红色表示上调的差异基因,绿色表示下调,白色的表示没有差异基因成功注释。 如果自己期望的一些基因没有成功注释,就代表没有吗?其实也不是,可以再试试KEGG官网数据库,使用BlastKOALA,上传蛋白序列-选择物种-填写邮箱
GSEA
Gene Set Enrichment Analysis 基因集富集分析,用于评估一个基因集的基因在表型相关度排序中的分布趋势,进而判断它们对表型的贡献
特点:
第一,它与GO和KEGG在算法层面上就是不同的,它采用的是FCS算法;
第二,与GO不同的地方在于,GO是先筛选基因(需要人为设定阈值),再判断差异基因在哪些通路有富集;GSEA可以考虑那些表达差异不大却功能重要的基因对通路影响,相比GO和KEGG能保留更多信息;
比如,你现在要研究某种昆虫的抗性,用传统的富集分析发现10个基因在细胞凋亡通路里有显著差异,同时还有10个基因在细胞扩增通路中有显著差异,那么到底是由于阻遏抗性的细胞凋亡引起抗性还是由于本身突变的细胞扩增产生了抗性? 用GSEA分析就可能会发现在细胞凋亡通路里,除了10个差异显著的基因表达量上升外,还有一些其他的相关基因也有整体上升的趋势;而扩增通路就不存在,因此可以进一步说明抗性和细胞凋亡的相关性更强
第三,研究表型的差异就要找到相应的基因,但是“孤木不成林”,即便是有一两个差异较大的基因,也不会直接导致表型变化。相对应的还要有一大帮相关的上游或者下游基因的变化,但它们的强度可能没有主效的那几个基因大。用GSEA就可以全面地进行分析,分析基因集中整体表达量得到的结果更可靠;
第四,你可能会想,为什么这个东西这么好,但是日常研究见不到呢?因为GSEA的数据库还并不完善,目前主要支持人类相关的研究,动植物还欠缺http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C3
原理: 使用一个排序的基因列表genelist(使用gseKEGG函数需要降序排列)、一个基因集geneset(比如可以自己选择某个代谢通路的基因、同一个GO注释的基因),判断geneset中的基因在genelist中是随机分布还是聚集在genelist的顶部或底部【落在顶部或底部表示geneset中的基因对表型差异用贡献】
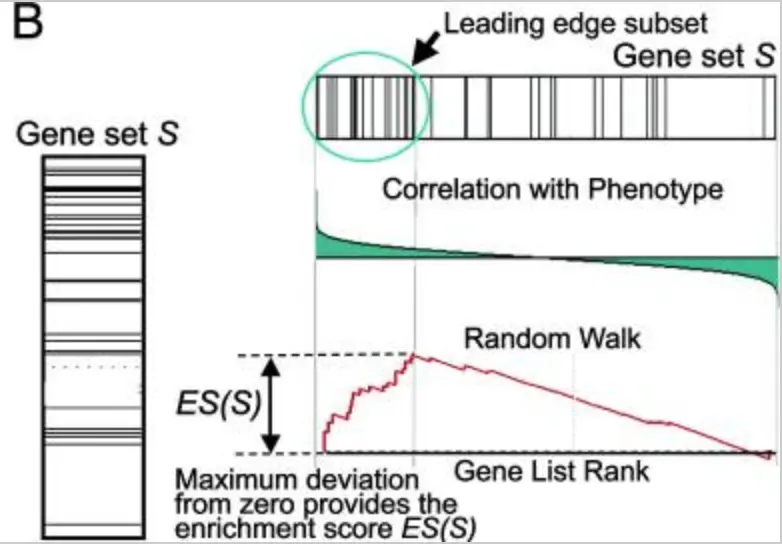
图片来自文章: 10.1073/pnas.0506580102
- ES是Enrichment score 富集得分:表示基因集的基因在基因列表两端的富集程度。开始从左到右基因集的第一个基因开始,每次计算统计值并且逐次累加,有一个在基因列表中的基因,就增加计算的统计值,反之就减小。并且每次的增加和减小都被记录在上图红线的位置,它代表了基因和表型的相关性。结果得到的峰值计作富集得分ES,正值表示基因集的基因主要在基因列表顶部富集,负值表示在底部富集

- 绿色圆圈表示的Leading edge subset:表示对ES富集得分最大的基因集中的基因,ES为正值表示是峰左侧的基因,负值表示右侧的基因。这个图中的每一条线表示基因集中一个基因
- ES是Enrichment score 富集得分:表示基因集的基因在基因列表两端的富集程度。开始从左到右基因集的第一个基因开始,每次计算统计值并且逐次累加,有一个在基因列表中的基因,就增加计算的统计值,反之就减小。并且每次的增加和减小都被记录在上图红线的位置,它代表了基因和表型的相关性。结果得到的峰值计作富集得分ES,正值表示基因集的基因主要在基因列表顶部富集,负值表示在底部富集