253-RNASeq表达量的各种M的统计方法,把你绕晕了吗?
刘小泽写于2021.8.24 今天花花问我一个问题:CPM和RPM是一样的吗?拿到RPM后,是不是可以和CPM的下游处理一样操作?为了给花花解决这个问题👩❤️👨,我花了3个小时搜集、整理、写代码、写推文🥳 确实,很多小伙伴对于各种M的统计指标表示分不清楚
因此今天我会通过文字+图片+公式+举例+代码的形式来说明问题
首先来看错综复杂的各种M统计指标
来自https://www.reneshbedre.com/blog/expression_units.html

最常见的莫过于:RPKM/FPKM,TPM,CPM了,除了它们还有很多比如RPM、TMM等
为何要存在这么多统计指标?
不同的统计学者有不同的认识,都想将自己的观点作为业界标准,所以才有了百花齐放的局面
我们要知道,我们拿到的RNASeq表达矩阵中,基因表达量数值并不是真实的情况,试想:
**情形1:**样本A的基因G1采用了20M的文库测序,而样本B的基因G1仅仅采用了10M的文库测序。多测多得,那么我们就不好判断表达矩阵中基因G1在A和B的表达量差异是不是由这个因素导致
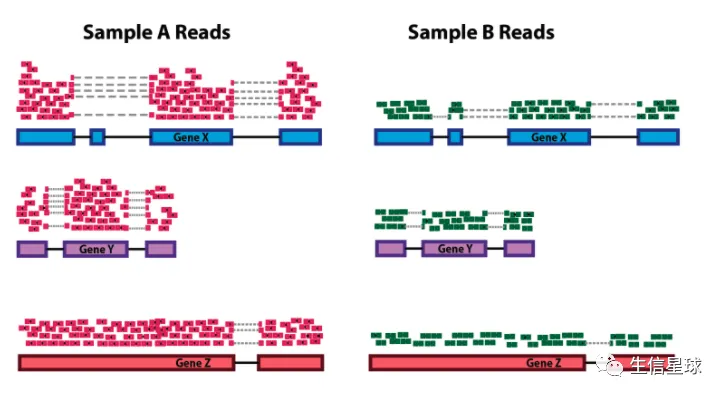
**情形2: ** 基因G1的长度是10K,而基因G2的长度是20K,那么不管在哪个样本中,测序过程中落在G2的概率都要比G1大,那么我们就不好判断表达矩阵中某样本基因G1和基因G2的差异是不是由这个因素导致
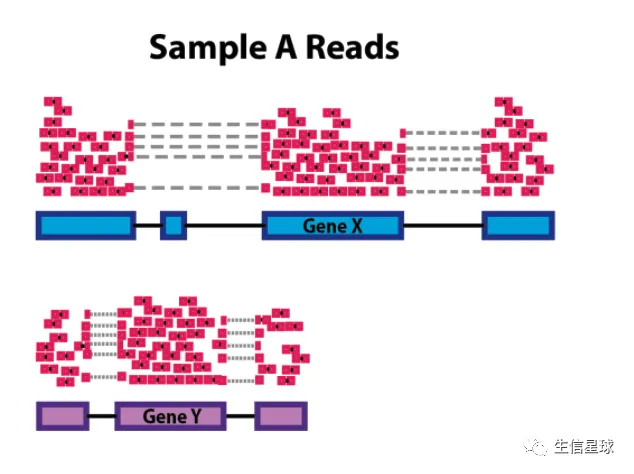
也就是说,RNASeq中的基因表达量主要受两方面因素的影响:文库大小和基因长度(不考虑实验中的人为技术误差)。这其中有的指标是只考虑了文库大小;有的既考虑了文库大小,又考虑了基因长度,因此才有了这么多的指标。
接下来一个一个指标看
指标1:RPM or CPM
RPM全称:Reads per million mapped reads;CPM全称: Counts per million mapped reads)
计算公式
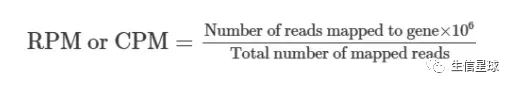
举例
一个样本测了5M reads(即文库大小为5M),其中有4M比对上了基因组,只有5000 reads比对到了基因G1,那么该基因的CPM/RPM值为:
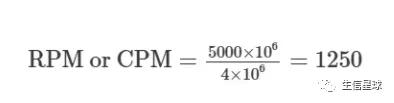
note
- 这个指标不考虑基因长度
代码
# 首先模拟一个数据(之后的演示都基于这个数据mat)
set.seed(123)
mat=matrix(sample(200:300,36,replace=T),6,6)
rownames(mat)=paste0('gene',1:6)
colnames(matz)=paste0('sample',1:6)
mat[5,]=sample(1:10,6,replace = T)
mat[6,]=sample(80:100,6,replace = T)
> mat
sample1 sample2 sample3 sample4 sample5 sample6
gene1 230 249 290 292 208 275
gene2 278 242 268 298 282 214
gene3 250 300 290 271 235 231
gene4 213 213 256 225 277 206
gene5 10 7 5 7 5 6
gene6 81 84 87 91 92 97
# 手动计算gene1在sample1的cpm
> colSums(mat)
sample1 sample2 sample3 sample4 sample5 sample6
1062 1095 1196 1184 1099 1029
> 230/colSums(mat)[1]*10^6
sample1
216572.5
# edger的cpm函数计算
> edgeR::cpm(mat)[1,1]
[1] 216572.5
> edgeR::cpm(mat) %>% colSums()
sample1 sample2 sample3 sample4 sample5 sample6
1e+06 1e+06 1e+06 1e+06 1e+06 1e+06
指标2:RPKM
全称:Reads per kilo base per million mapped reads
当然,还有一个FPKM (Fragments per kilo base per million mapped reads),更合适用于PE 的RNASeq。因为PE reads在比对过程中,可能两条reads同时比对,也有可能仅有一条read比对到参考基因组,如果按RPKM,两条reads同时比对这种情况,统计结果就是2,但实际上这2条reads仍然属于同一个fragment,仅仅是方向不同而已,所以不管是比对上1条还是2条reads,用FPKM的结果都是1
计算公式

10^3 normalizes for gene length and 10^6 for sequencing depth factor
举例
一个样本单端文库测了5M reads(即单端文库大小为5M),其中有4M比对上了基因组,只有5000 reads比对到了基因G1(长度是2kb),那么该基因的RPKM值为:
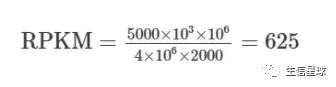
note
- RPKM和FPKM增加了基因长度的考量
- 小tip:**或许叫做(RPMK/FPMK)更有帮助记忆?**因为先处理文库大小(所以最后两个字母M在前),再处理基因长度(所以K在后)
代码
# 假设拿到基因长度
lengths <- c(1000,2000,500,1500,3000,2500)
# 各个样本文库大小
> colSums(mat)
sample1 sample2 sample3 sample4 sample5 sample6
1062 1095 1196 1184 1099 1029
# RPKM需要先对文库标准化(以第一列为例演示)
x1 = mat[,1]*10^6/colSums(mat)[1]
# 再对长度标准化
> x1*10^3/lengths
gene1 gene2 gene3 gene4 gene5 gene6
216572.505 130885.122 470809.793 133709.981 3138.732 30508.475
# 写成函数就是:
countToFpkm <- function(x){
N <- sum(x)
x*10^3*10^6/(N*lengths)
}
fpkm <- apply(mat,2,countToFpkm)
> fpkm[1:4,1:4]
sample1 sample2 sample3 sample4
gene1 216572.5 227397.3 242474.9 246621.6
gene2 130885.1 110502.3 112040.1 125844.6
gene3 470809.8 547945.2 484949.8 457770.3
gene4 133710.0 129680.4 142697.9 126689.2
> colSums(fpkm)
sample1 sample2 sample3 sample4 sample5 sample6
985624.6 1048340.9 1012653.3 989639.6 948256.0 993326.9
注意最后的colSums(fpkm)需要注意,等下这里会和TPM进行区分,从而看出二者明显的差异!
指标3:TPM
全称是:Transcripts per million
计算公式

这里直接用代码解释它和RPKM的不同吧:
代码
# 以第一列为例
# 先对长度标准化
x1 = mat[,1]*10^3/lengths
# 再对文库标准化
> x1*10^6/sum(x1)
gene1 gene2 gene3 gene4 gene5 gene6
219731.227 132794.090 477676.581 135660.149 3184.511 30953.442
# 写成函数就是:
countToTpm <- function(x){
normLen <- x*10^3/lengths
normLen*10^6/sum(normLen)
}
tpm=apply(mat, 2, countToTpm)
> tpm[1:4,1:4]
sample1 sample2 sample3 sample4
gene1 219731.2 216911.6 239445.1 249203.5
gene2 132794.1 105406.8 110640.2 127162.0
gene3 477676.6 522678.4 478890.3 462562.6
gene4 135660.1 123700.6 140914.8 128015.5
> colSums(tpm)
sample1 sample2 sample3 sample4 sample5 sample6
1e+06 1e+06 1e+06 1e+06 1e+06 1e+06
可以看到,TPM标准化后的文库大小,各个样本是一致的,也实现了去除文库和基因长度差异后,没有引入新的文库差异的初衷
note
我之前也做的一张图,对比了TPM和FPKM
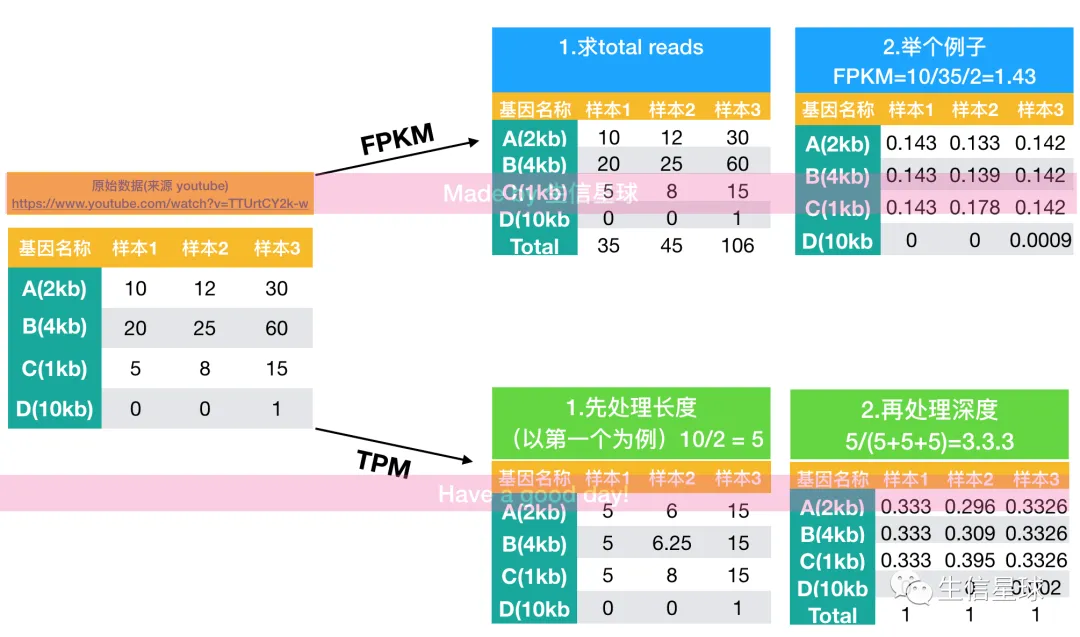
可以看到无论是RPKM还是TPM,都有一个重点就是基因长度。如果你对基因长度计算感兴趣,可以看最后的补充阅读部分(RPKM概念及计算方法)
指标4:TMM
全称是:Trimmed Mean of M-values,主要存在于差异分析中
和之前的TPM、RPKM不同,它着眼于样本间的比较,而不是样本内部比较。它基于一个假设:大部分的基因并非差异表达
- Normalize the total RNA output among the samples and does not consider gene length or library size for normalization
- TMM的计算可以借助edgeR。It does not consider gene length for normalization as it assumes that the gene length would be constant between the samples
计算公式
- 计算每个样本中每个基因的library size normalized read count
- 然后计算log2 fold change between the two samples (M value)

- 计算absolute expression count (A value)
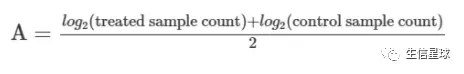
- double trim the upper and lower percentages of the data (trim M values by 30% and A values by 5%)
- Get weighted mean of M
代码
# 需要制定一个分组
group <- factor(c('c','c', 'c', 't', 't', 't'))
library(edgeR)
y <- DGEList(counts=mat, group=group)
# normalize for library size by cacluating scaling factor using TMM (default method)
y <- calcNormFactors(y)
# 可以看到每个样本根据各自文库大小,计算了norm.factors
> y
An object of class "DGEList"
$counts
Sample1 Sample2 Sample3 Sample4 Sample5 Sample6
gene1 276 213 250 217 262 233
gene2 258 261 267 293 272 288
gene3 286 228 257 294 240 300
gene4 261 210 257 223 271 294
gene5 4 5 9 8 1 9
gene6 92 99 97 91 98 80
$samples
group lib.size norm.factors
Sample1 c 1177 0.9880091
Sample2 c 1016 1.0158008
Sample3 c 1137 0.9877519
Sample4 t 1126 1.0290219
Sample5 t 1144 0.9829446
Sample6 t 1204 0.9973071
#Obtain the TMM values of your data
> cpm(y)
sample1 sample2 sample3 sample4 sample5 sample6
gene1 209788.154 231597.913 241569.42 247519.357 195093.769 262820.562
gene2 253570.030 225087.128 223243.47 252605.371 264502.130 204522.183
gene3 228030.603 279033.630 241569.42 229718.307 220418.441 220769.272
gene4 194282.073 198113.877 213247.49 190725.532 259812.376 196876.494
gene5 9121.224 6510.785 4164.99 5933.683 4689.754 5734.267
gene6 73881.915 78129.416 72470.83 77137.882 86291.475 92703.980
可以看到,和第一个CPM的计算结果还是不同的,毕竟这里加入了分组比较(treat vs control)才能计算TMM最后的那个字母M(M-value)
指标5:DESeq median-of-ratios
不管是DESeq还是DESeq2,校正方法都和TMM类似,同样假设:大部分基因并非差异表达
同样不考虑基因长度,关注点就是一个基因在不同的样本差异
所以重点就是size factor的计算
size factor的计算又依赖于计算median of the ratios of observed counts
- 简而言之,size factor首先要将每个样本的所有基因表达量(raw count)除以该样本中表达量的几何平均数(geometric mean);然后取中位数(median),作为该样本的size factor;最后利用每个样本的size factor去进行标准化
代码
# 构建一个样本比较矩阵
cond = data.frame(sample = colnames(mat),
condition = group)
> cond
sample condition
1 sample1 c
2 sample2 c
3 sample3 c
4 sample4 t
5 sample5 t
6 sample6 t
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = mat,
colData = cond,
design = ~ condition)
> dds
class: DESeqDataSet
dim: 6 6
metadata(1): version
assays(1): counts
rownames(6): gene1 gene2 ... gene5 gene6
rowData names(0):
colnames(6): sample1 sample2 ... sample5 sample6
colData names(2): sample condition
dds <- estimateSizeFactors(dds)
y = counts(dds, normalized = TRUE)
> y
sample1 sample2 sample3 sample4 sample5 sample6
gene1 244.59887 258.846568 272.35173 275.824071 215.221126 301.977645
gene2 295.64560 251.569757 251.69057 281.491689 291.790180 234.993513
gene3 265.86834 311.863335 272.35173 255.987408 243.158484 253.661221
gene4 226.51983 221.422968 240.42084 212.535671 286.616596 226.208708
gene5 10.63473 7.276811 4.69572 6.612221 5.173585 6.588603
gene6 86.14134 87.321734 81.70552 85.958871 95.193960 106.515751
> sizeFactors(dds)
sample1 sample2 sample3 sample4 sample5 sample6
0.9403150 0.9619598 1.0647995 1.0586458 0.9664479 0.9106634
note
- 有人说DESeq or DESeq2 performs better for between-samples comparisons,对此你怎么看?
- 如果是RSEM的结果,推荐使用
tximport导入,然后用DESeqDataSetFromTximport()for 衔接DESeq2的差异分析 - 当然,将RSEM的结果四舍五入也是可以的,但效果相比于
tximport要差一点
指标6:GeTMM
全称是:Gene length corrected TMM
Smid et al., 2018在BMC Bioinformatics发表了这个指标,特色是兼容样本内+样本间的比较
- 基于TMM,但允许加入基因长度的校正(计算RPK,即 reads per Kbp of gene length)
- 首先基于raw count计算每个基因的RPK,然后进行TMM校正
代码
# calculate reads per Kbp of gene length (corrected for gene length)
# gene length is in bp in exppression dataset and converted to Kbp
rpk <- ( (mat*10^3 )/lengths)
y <- DGEList(counts=rpk, group=group)
y <- calcNormFactors(y)
# count per million read (normalized count)
norm_counts <- cpm(y)
> norm_counts
sample1 sample2 sample3 sample4 sample5 sample6
gene1 219727.527 212302.529 243145.327 252466.701 205958.835 258946.274
gene2 132791.853 103167.092 112349.910 128827.186 139616.325 100753.641
gene3 477668.538 511572.359 486290.654 468619.698 465387.751 435029.740
gene4 135657.865 121072.125 143092.422 129691.798 182854.478 129316.200
gene5 3184.457 1989.448 1397.387 2017.428 1650.311 1883.246
gene6 30952.921 28648.052 29177.439 31471.876 36438.871 36534.965
讨论
http://luisvalesilva.com/datasimple/rna-seq_units.html 提出:样本间比较用CPM;样本内比较TPM优于RPKM
CPM is an appropriate unit for inter-sample comparison.
When comparing feature expression within samples, TPM should be used instead of RPKM/FPKM
补充阅读
公众号内需要阅读原文才能打开下面👇链接哦