239-如何统计bam文件的覆盖度和测序深度
刘小泽写于2021.04.12 这次的目的有两个:一个是想看bam文件在全基因或者某些区域的覆盖度;另一个是想看在某些区域的测序深度
首先来看两个名词
这两个名词是相辅相成的,一个看横向,一个看纵向

横向——覆盖度(coverage)
所谓覆盖度,也可以理解成“有多少基因组区域被覆盖”,一般是一个百分比。
比如我们的bam文件在chr1的覆盖度为70%,就表示只有30%的chr1范围内,一条reads也没有
纵向——测序深度(depth)
测序深度是统计落在基因组区域中的总碱基数量,然后除以这个区域的长度
结果一般是1X、5X、10X这样表示
看到这个网站给出的解释不错:https://www.ecseq.com/support/ngs/how-to-calculate-the-coverage-for-a-sequencing-experiment

如果看整个区域(112nt):
# 覆盖度
(46nt - 2nt) / 112nt = 39.3%
# 测序深度(总共188个nt比对上)
188/112nt=1.68 fold
如果看某个区域(46nt):
# 覆盖度
(46nt - 2nt) / 46nt = 95.7%
# 测序深度(总共188个nt比对上)
188/46=4.09 fold
如果只看某个位点(比如红色的G对应的位点):
# 覆盖度
6 fold
覆盖度多少是好呢?
There is no general guideline for determining the optimal coverage for a sequencing project.
It highly depends on the type of experiment, the species, the input material, the sequencing platform and other factors.
这个网站推荐了一些常规的NGS测序深度参考:https://genohub.com/recommended-sequencing-coverage-by-application/
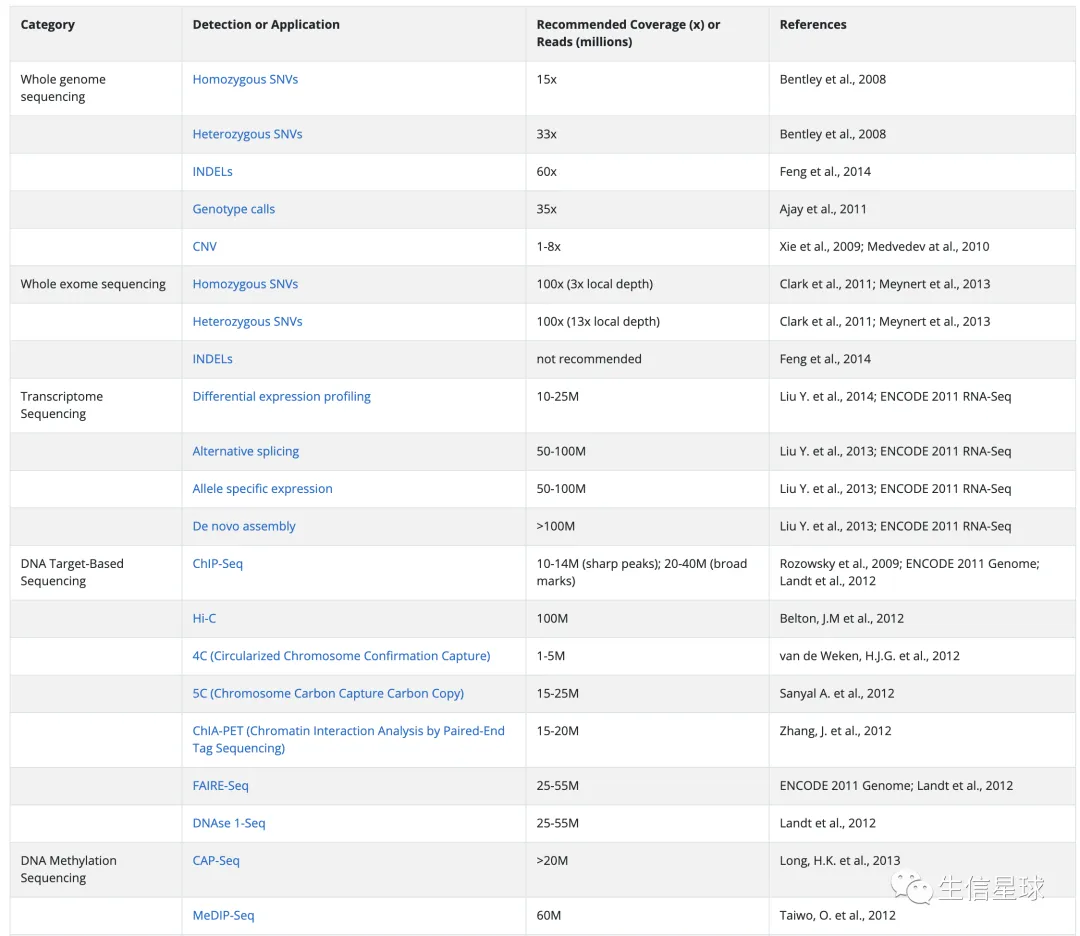
使用bamdst计算覆盖度
安装
git clone https://github.com/shiquan/bamdst.git
cd bamdst/
make
./bamdst -h
使用
需要制作一个bed文件
# tab分隔
cat >test.bed
chr6 52156548 52161782
然后
bamdst -p test.bed -o ./ test.bam
# -o:output dir
结果
cat chromosomes.report
#Chromosome DATA(%) Avg depth Median Coverage% Cov 4x % Cov 10x % Cov 30x % Cov 100x %
chr6 100.00 0.15 0.0 8.90 0.00 0.00 0.00 0.00
结合IGV看一下结果

上面统计的1X 覆盖率是8.9%,也就是计算了这三段的长度,然后除以区域长度即52161782-52156548=5234
使用samtools depth计算测序深度
统计某个染色体的平均深度
小鼠染色体长度:http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/mm10.chrom.sizes
人染色体长度:http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.chrom.sizes
# 统计某个染色体的平均深度
grep -vi 'random' mm10.chrom.sizes | grep -vi un | while read i;do
conf=($i)
chr=${conf[0]}
len=${conf[1]}
samtools depth -r $chr test.bam | awk -v chr=$chr -v len=$len '{sum+=$3} END { print chr,"-Average = ",sum/len}';done
# chr1 -Average = 1.01466
# chr2 -Average = 1.06905
统计某个染色体上的某个区域的平均深度
# 指定区域
chr=chr6 && start=52156548 && end=52161782 && samtools depth -r $chr:$start-$end test.bam | awk -v start=$start -v end=$end '{sum+=$3} END { print "Average = ",sum/(end-start)}'
# Average = 0.151891
这个结果和上面bamdst统计的0.15一致