234-给你几条蛋白序列,用R语言怎么比对?
刘小泽写于2021.2.2 今天我们以两条蛋白序列为例,来看看用R怎么比对,并且重点是要找到目标位点的比对结果。当然,今天这个R包不仅可以比对蛋白序列,还支持DNA, RNA
安装R包
rm(list=ls())
options(stringsAsFactors = F)
# BiocManager::install("msa")
suppressMessages(library(msa))
library(stringr)
下载蛋白序列
首页(https://www.uniprot.org/)输入:基因名_物种,获取ID号
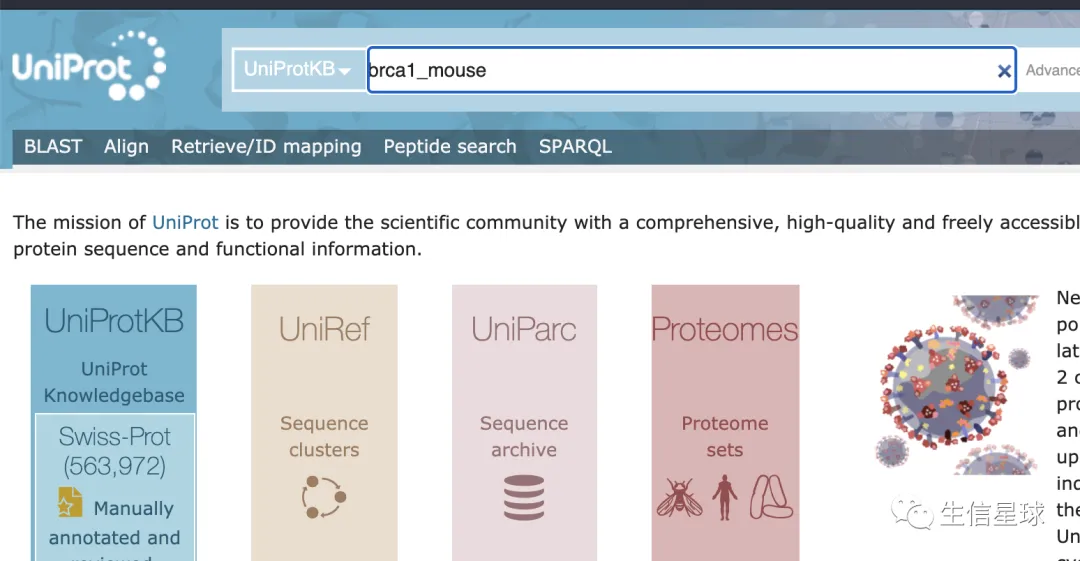

然后就可以下载到小鼠该基因的蛋白序列
注意网页链接的规律
https://www.uniprot.org/uniprot/P48754.fasta
同理,我们也获取到human的该基因蛋白序列: https://www.uniprot.org/uniprot/P38398.fasta
R中继续操作
官方文档在:https://bioconductor.org/packages/release/bioc/vignettes/msa/inst/doc/msa.pdf
# 读取序列
hg <- readAAStringSet('P38398.fasta')
mm <- readAAStringSet('P48754.fasta')
# 结果保存为AAStringSet对象。当然,还支持读入 DNAStringSet, and RNAStringSet对象

# 进行比对
mySequences=c(mm,hg)
myFirstAlignment <- msa(mySequences)
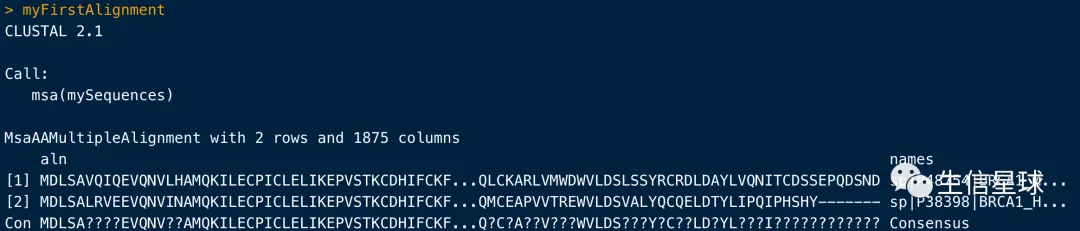
可以查看全部比对结果
print(myFirstAlignment, show="complete")

还可以输出为pdf
msaPrettyPrint(myFirstAlignment, output="pdf", showNames="none",showLogo="none", askForOverwrite=FALSE, verbose=FALSE)
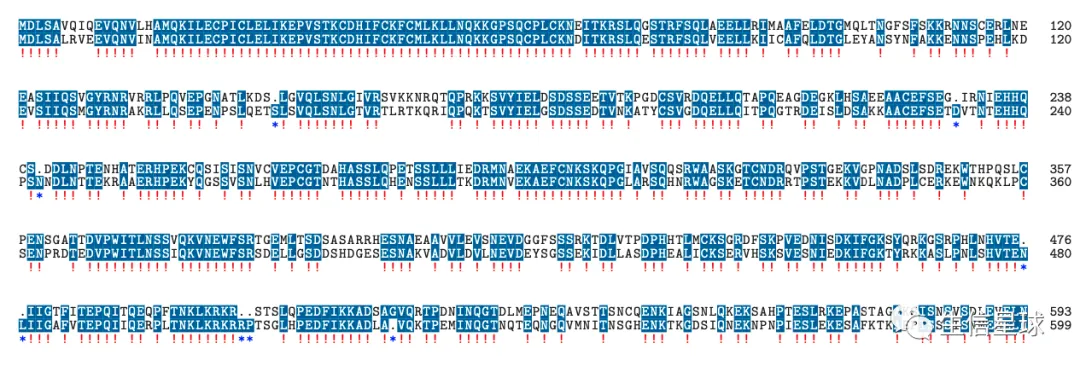
其中感叹号!就是全部比对上的,*就是存在gap的位点
当然,如果比对的序列很多,还可以增加logo
# 示例代码
msaPrettyPrint(myFirstAlignment, output="asis", y=c(164, 213),subset=c(1:6), showNames="none", showLogo="top",logoColors="rasmol", shadingMode="similar",showLegend=FALSE, askForOverwrite=FALSE)

接下来才是重点
上面函数使用很简单,关键是我们得到了比对的结果,怎么提取我想要的位点呢
其实之前biostar上也有人提出了这个问题:https://www.biostars.org/p/467870/
也就是说,他之前感兴趣的位点是82位的AA,想要看看比对后这个AA对应着哪个AA。但是他发现,比对后原来的AA位置变成了95。所以不知道应该怎么去统计
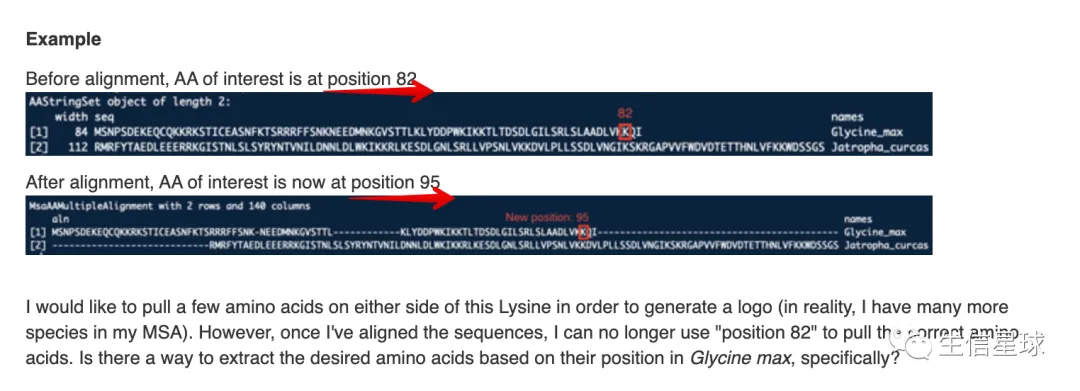
这里给出我自己的理解
**首先明确,比对前后的差异在什么地方?**=》原来可以比对的当然能对应,但是不能比对的分了两种情况:一个是硬比对(比如D比对到了N);一个是软比对,即增加了gap(比如D 比对到. )。
麻烦就出在gap身上,原来序列没有,比对过程加上去。
因此,我们在统计的时候,就需要考虑进来gap的数量
这里以小鼠为query,人为target。也就是说,小鼠的位点可以按照之前下载的序列,但是人的位点,就需要加上gap。
以下面👇这个位点Q为例

这里显示的一条序列长度是120,正因为混入了两个gap(蓝色的星号表示),所以原本238的位置,现在处于240【所以,比对结果右侧数字记录的就是:原始的位点所处的位置】
我们可以轻松得到小鼠原来第238位的位点:
> pos=238 > str_sub(as.character(mm),pos,pos) [1] "Q"那human呢?用同样的方法能行吗?
> str_sub(as.character(hg),pos,pos) [1] "H"结果显然不对,它应该是
Q,但这里显示H。因此,我们应该把gap考虑进来在小鼠的第238位点之前,一共有几个gap?
# 这里需要从比对结果(myFirstAlignment@unmasked)中提取,即myFirstAlignment@unmasked[1] > ngap=str_sub(as.character(myFirstAlignment@unmasked[1]),1,pos) %>% str_count('-') > ngap [1] 2这2个gap也符合我们的肉眼验证,因此human此时对应的位置应该是:
> hg_site=str_sub(as.character(myFirstAlignment@unmasked[2]),pos+ngap,pos+ngap) > hg_site [1] "Q"
原以为这样就结束了,其实并没有
我测试了好多个,结果都对,但又随机挑选了一个502位点,发现了错误:
正确的应该是:S<=>T,但我得到的是:S<=>R

> pos=502
# 小鼠的
> str_sub(as.character(mm),pos,pos)
[1] "S"
# 人的
> ngap=str_sub(as.character(myFirstAlignment@unmasked[1]),1,pos+ngap)%>% str_count('-')
> str_sub(as.character(myFirstAlignment@unmasked[2]),pos+ngap,pos+ngap)
[1] "R"
错误原因就是ngap计算错误:
# 此时计算的是
> ngap
# 但其实数一数,在502位点前,其实还有2个,因此是7个
但为什么gap会统计错误呢?
因为我们这里给出的pos=502,在比对结果中,是落在了真实502位置的前面,而且恰巧也落在了那2个新的gap的前面,所以没有统计上。这里我想了一种解决方案,就是增加一步while循环,来探索在比对结果的502位点之后,有没有新的gap出现
ngap=str_sub(as.character(myFirstAlignment@unmasked[1]),1,pos) %>% str_count('-')
new_ngap=str_sub(as.character(myFirstAlignment@unmasked[1]),1,pos+ngap) %>% str_count('-')
while (new_ngap != ngap) {
ngap=new_ngap
next
}
mm_site=str_sub(as.character(mm),pos,pos)
hg_site=str_sub(as.character(myFirstAlignment@unmasked[2]),pos+ngap,pos+ngap)