226-以后看NCBI,再也不用鼠标点点点了
刘小泽写于2021.1.11
有用的教程
rentrez包简介
NCBI是经常使用到的,如果想更快去获取数据,首先想到下载NCBi的数据库,但实际上这是不可行的。一方面是因为它经常更新,另一方面感受一下它的数据量大小。
截止到2020-11-11,NCBI就有:
31.7 million papers in PubMed
6.6 million full-text records available in PubMed Central
The NCBI Nucleotide Database (which includes GenBank) has data for 432 million different sequences
dbSNP describes 702 million different genetic variants
1.86 million species in the NCBI taxonomy
27 thousand disease-associated records in OMIM
因此,更有效率更有时效性的办法是:使用NCBI网站提供的API接口——Entrez Utilities (Eutils for short)。在R中,这个工具可以用rentrez,进行在线获取数据
0-rentrez使用起步
全部数据库类型
要想用好NCBI的API工具,起码要知道NCBI数据库都包含哪些类型数据吧
library(rentrez)
entrez_dbs()
## [1] "pubmed" "protein" "nuccore"
## [4] "ipg" "nucleotide" "structure"
## [7] "sparcle" "protfam" "genome"
## [10] "annotinfo" "assembly" "bioproject"
## [13] "biosample" "blastdbinfo" "books"
## [16] "cdd" "clinvar" "gap"
## [19] "gapplus" "grasp" "dbvar"
## [22] "gene" "gds" "geoprofiles"
## [25] "homologene" "medgen" "mesh"
## [28] "ncbisearch" "nlmcatalog" "omim"
## [31] "orgtrack" "pmc" "popset"
## [34] "proteinclusters" "pcassay" "biosystems"
## [37] "pccompound" "pcsubstance" "seqannot"
## [40] "snp" "sra" "taxonomy"
## [43] "biocollections" "gtr"
知道了有这些数据库,然后这个包有3个十分常用的函数:

检索某个数据库
# 看基本信息
entrez_db_summary("pubmed")
## DbName: pubmed
## MenuName: PubMed
## Description: PubMed bibliographic record
## DbBuild: Build210110-1313.1
## Count: 31990870
## LastUpdate: 2021/01/10 17:45
# 看某个数据库的检索关键词
entrez_db_searchable("sra")
## Searchable fields for database 'sra'
## ALL All terms from all searchable fields
## UID Unique number assigned to publication
## FILT Limits the records
## ACCN Accession number of sequence
## TITL Words in definition line
## PROP Classification by source qualifiers and molecule type
## WORD Free text associated with record
## ORGN Scientific and common names of organism, and all higher levels of taxonomy
## AUTH Author(s) of publication
## PDAT Date sequence added to GenBank
## MDAT Date of last update
## GPRJ BioProject
## BSPL BioSample
## PLAT Platform
## STRA Strategy
## SRC Source
## SEL Selection
## LAY Layout
## RLEN Percent of aligned reads
## ACS Access is public or controlled
## ALN Percent of aligned reads
## MBS Size in megabases
1-ID搜索(search)
基础搜索
搜索功能是使用频率最高的功能,我们可以设置database name (db)、search term (term) 进行简单搜索
r_search <- entrez_search(db="pubmed", term="single cell[All Fields]")
r_search
## Entrez search result with 49388 hits (object contains 20 IDs and no web_history object)
## Search term (as translated): single cell[All Fields]
发现hits比返回的ID数量多,这是因为默认参数retmax=20。当然,如果需要返回的数量非常多,就不需要设置这个参数了,而是通过设置entrez_fetch中的web_history(后面会提到)
another_r_search <- entrez_search(db="pubmed", term="R Language", retmax=40)
another_r_search
## Entrez search result with 14122 hits (object contains 40 IDs and no web_history object)
## Search term (as translated): R[All Fields] AND ("programming languages"[MeSH Te ...
返回的结果中ID最为重要,可以后面得到summary data或者与其他数据库联动
r_search$ids
## [1] "33421851" "33421727" "33421713" "33421512" "33421413"
## [6] "33420958" "33420907" "33420693" "33420657" "33420491"
## [11] "33420489" "33420488" "33420427" "33420425" "33420268"
## [16] "33420144" "33420075" "33419925" "33419885" "33419769"
设置搜索条件
搜索条件可以是query[SEARCH FIELD],或者AND, OR and NOT ,例如:
entrez_search(db="sra",
term="Homo sapiens[ORGN]",
retmax=0)
## Entrez search result with 2358175 hits (object contains 0 IDs and no web_history object)
## Search term (as translated): "Homo sapiens"[Organism]
# 加日期限定
entrez_search(db="sra",
term="Homo sapiens[ORGN] AND 2019:2020[PDAT]",
retmax=0)
## Entrez search result with 984743 hits (object contains 0 IDs and no web_history object)
## Search term (as translated): "Homo sapiens"[Organism] AND 2019[PDAT] : 2020[PDA ...
# 添加其他物种
entrez_search(db="sra",
term="(Homo sapiens[ORGN] OR Mus musculus[ORGN]) AND 2019:2020[PDAT]",
retmax=0)
## Entrez search result with 1664051 hits (object contains 0 IDs and no web_history object)
## Search term (as translated): ("Homo sapiens"[Organism] OR "Mus musculus"[Organi ...
其中的SEARCH FIELD依然是看entrez_db_searchable("sra")返回的结果
搜索文献
比如拿到一篇文章的DOI号,如何快速获得它的pubmed ID呢?
hox_paper <- entrez_search(db="pubmed", term="10.1038/nature08789[doi]")
hox_paper$ids
## [1] "20203609"
将搜索结果可视化
上面看到entrez_search()返回的结果包含了一些数量信息,我们可以提取进行作图。
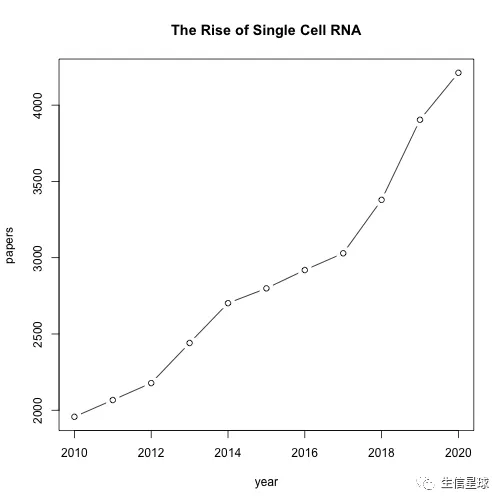
比如粗略地看10年间单细胞转录组的发展速度
papers_by_year <- function(years, search_term){
return(sapply(years, function(y) entrez_search(db="pubmed",term=search_term, mindate=y, maxdate=y, retmax=0)$count))
}
year <- 2010:2020
papers <- sapply(year, papers_by_year, search_term="single cell RNA", USE.NAMES=FALSE)
plot(year, papers, type='b', main="The Rise of Single Cell RNA")
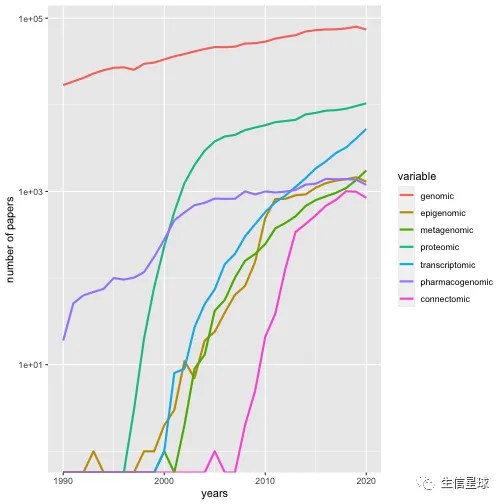
另外看看多组学的发展
years <- 1990:2020
total_papers <- papers_by_year(years, "")
omics <- c("genomic", "epigenomic", "metagenomic", "proteomic", "transcriptomic", "pharmacogenomic", "connectomic" )
trend_data <- sapply(omics, function(t) papers_by_year(years, t))
trend_props <- trend_data/total_papers
library(reshape)
library(ggplot2)
trend_df <- melt(data.frame(years, trend_data), id.vars="years")
p <- ggplot(trend_df, aes(years, value, colour=variable))
p + geom_line(size=1) + scale_y_log10("number of papers")
2-ID关联(link)
NCBI另一个强大之处在于:各个数据库的关联性很强,基本上输入一个ID,得到的结果满满全是链接。为了展示这种强大,这里举一个例子:Amyloid Beta Precursor gene(NCBI 的gene ID是351),与阿尔茨海默病患者大脑中形成的斑块有关
关联所有可能的数据库
all_the_links <- entrez_link(dbfrom='gene', id=351, db='all')
names(all_the_links)
## [1] "links" "file"
all_the_links$links
## elink result with information from 57 databases:
## [1] gene_bioconcepts
## [2] gene_biosystems
## [3] gene_biosystems_all
## [4] gene_clinvar
## [5] gene_clinvar_specific
## [6] gene_dbvar
## [7] gene_genome
## [8] gene_gtr
## [9] gene_homologene
## [10] gene_medgen_diseases
## [11] gene_pcassay_alltarget_list
## [12] gene_pcassay_alltarget_summary
## [13] gene_pcassay_rnai
## [14] gene_pcassay_rnai_active
## [15] gene_pcassay_target
## [16] gene_probe
## [17] gene_snp
## [18] gene_structure
## [19] gene_bioproject
## [20] gene_cdd
## [21] gene_gene_h3k4me3
## [22] gene_gene_neighbors
## [23] gene_genereviews
## [24] gene_genome2
## [25] gene_geoprofiles
## [26] gene_nuccore
## [27] gene_nuccore_mgc
## [28] gene_nuccore_pos
## [29] gene_nuccore_refseqgene
## [30] gene_nuccore_refseqrna
## [31] gene_nucest_clust
## [32] gene_nucleotide
## [33] gene_nucleotide_clust
## [34] gene_nucleotide_mgc
## [35] gene_nucleotide_mgc_url
## [36] gene_nucleotide_pos
## [37] gene_omim
## [38] gene_pcassay_proteintarget
## [39] gene_pccompound
## [40] gene_pcsubstance
## [41] gene_pmc
## [42] gene_pmc_nucleotide
## [43] gene_protein
## [44] gene_protein_refseq
## [45] gene_pubmed
## [46] gene_pubmed_all
## [47] gene_pubmed_citedinomim
## [48] gene_pubmed_highlycited
## [49] gene_pubmed_latest
## [50] gene_pubmed_pmc_nucleotide
## [51] gene_pubmed_reviews
## [52] gene_pubmed_rif
## [53] gene_snp_geneview
## [54] gene_sparcle
## [55] gene_taxonomy
## [56] gene_unigene
## [57] gene_varview
结果的命名方式都是:[source_database]_[linked_database],因此想看到这个基因对应的PubMed ID,就可以:
length(all_the_links$links$gene_pmc)
## [1] 1241
all_the_links$links$gene_pmc[1:10]
## [1] "7656556" "7510992" "7490758" "7378830" "7296003"
## [6] "7291702" "7247290" "7113242" "6991300" "6956526"
或者想看这个基因在疾病中的作用,可以链接到clinVar中
clinVar是NCBI主办的与疾病相关的人类基因组变异数据库。它的强大在于整合了dbSNP、dbVar、Pubmed、OMIM等多个数据库在遗传变异和临床表型方面的数据信息,将变异、临床表型、实证数据以及功能注解与分析等四个方面的信息,通过专家评审,逐步形成一个标准的、可信的、稳定的遗传变异-临床表型相关的数据库
head(all_the_links$links$gene_clinvar)
## [1] "983157" "980228" "967074" "963922" "932452" "899123"
length(all_the_links$links$gene_clinvar)
## [1] 232
关联某个指定的数据库
还是以上面的351基因为例,如果我们目的是找与这个基因相关的转录本信息
Transcript sequences are stored in the nucleotide database (referred to as
nuccorein EUtils)
nuc_links <- entrez_link(dbfrom='gene', id=351, db='nuccore')
nuc_links
## elink object with contents:
## $links: IDs for linked records from NCBI
##
nuc_links$links
## elink result with information from 5 databases:
## [1] gene_nuccore gene_nuccore_mgc
## [3] gene_nuccore_pos gene_nuccore_refseqgene
## [5] gene_nuccore_refseqrna
nuc_links$links$gene_nuccore_refseqrna
## [1] "1889693417" "1869284273" "1676441520" "1676319912"
## [5] "1675178653" "1675118060" "1675113449" "1675055422"
## [9] "1674986144" "1519241754" "1370481385" "324021746"
有了这些ID之后,就可以利用entrez_fetch() or entrez_summary() 去获得更多内容【之后提到】
关联外部链接
entrez_link()除了可以内部关联,还可以继续关联外部数据库。
比如:找某些paper的外部链接
paper_links <- entrez_link(dbfrom="pubmed", id=25500142, cmd="llinks")
paper_links
## elink object with contents:
## $linkouts: links to external websites
paper_links$linkouts
## $ID_25500142
## $ID_25500142[[1]]
## Linkout from Elsevier Science
## $Url: https://linkinghub.elsevie ...
##
## $ID_25500142[[2]]
## Linkout from Europe PubMed Central
## $Url: http://europepmc.org/abstr ...
##
## $ID_25500142[[3]]
## Linkout from Ovid Technologies, Inc.
## $Url: http://ovidsp.ovid.com/ovi ...
##
## $ID_25500142[[4]]
## Linkout from PubMed Central
## $Url: https://www.ncbi.nlm.nih.g ...
##
## $ID_25500142[[5]]
## Linkout from MedlinePlus Health Information
## $Url: https://medlineplus.gov/al ...
##
## $ID_25500142[[6]]
## Linkout from Mouse Genome Informatics (MGI)
## $Url: http://www.informatics.jax ...
linkout_urls(paper_links)
## $ID_25500142
## [1] "https://linkinghub.elsevier.com/retrieve/pii/S0014-4886(14)00393-8"
## [2] "http://europepmc.org/abstract/MED/25500142"
## [3] "http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=linkout&SEARCH=25500142.ui"
## [4] "https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/25500142/"
## [5] "https://medlineplus.gov/alzheimersdisease.html"
## [6] "http://www.informatics.jax.org/reference/25500142"
当然,更多更强大的功能可以继续看fulltext manual:https://books.ropensci.org/fulltext/
多个ID的关联
上面所看到的都是将1个ID关联到其他数据库,但如果想关联多个,也是可以的,并且可以分别返回结果。
all_links_sep <- entrez_link(db="protein", dbfrom="gene", id=c("93100", "223646"), by_id=TRUE)
all_links_sep
## List of 2 elink objects,each containing
## $links: IDs for linked records from NCBI
##
lapply(all_links_sep, function(x) x$links$gene_protein)
## [[1]]
## [1] "1387845369" "1387845338" "1370513171" "1370513169"
## [5] "1034662000" "1034661998" "1034661996" "1034661994"
## [9] "1034661992" "558472750" "545685826" "194394158"
## [13] "166221824" "154936864" "122346659" "119602646"
## [17] "119602645" "119602644" "119602643" "119602642"
## [21] "37787309" "37787307" "37787305" "33991172"
## [25] "21619615" "10834676"
##
## [[2]]
## [1] "1720383952" "148697547" "148697546" "81899807"
## [5] "74215266" "74186774" "37787317" "37589273"
## [9] "31982089" "26339824" "26329351"
3-ID概况(summary)
前面介绍的entrez_search()以及entrez_link(),最后都可以获取一些ID。但具体这些ID的信息是什么,还需要继续探索。Eutils API提供了两种方法:
entrez_summary()返回比较精炼的结果(概况)entrez_fetch()返回最全的结果
看看entrez_summary()如何使用吧
all_the_links <- entrez_link(dbfrom='gene', id=351, db='all')
all_the_links$links$gene_pmc[1:10]
## [1] "7656556" "7510992" "7490758" "7378830" "7296003"
## [6] "7291702" "7247290" "7113242" "6991300" "6956526"
# 就以其中一个6956526为例
taxize_summ <- entrez_summary(db="pubmed", id=6956526)
taxize_summ
## esummary result with 42 items:
## [1] uid pubdate epubdate
## [4] source authors lastauthor
## [7] title sorttitle volume
## [10] issue pages lang
## [13] nlmuniqueid issn essn
## [16] pubtype recordstatus pubstatus
## [19] articleids history references
## [22] attributes pmcrefcount fulljournalname
## [25] elocationid doctype srccontriblist
## [28] booktitle medium edition
## [31] publisherlocation publishername srcdate
## [34] reportnumber availablefromurl locationlabel
## [37] doccontriblist docdate bookname
## [40] chapter sortpubdate sortfirstauthor
entrez_summary()返回的结果也是类似列表,利用$提取
# 将pubmed ID转为其他ID
taxize_summ$articleids
## idtype idtypen value
## 1 pubmed 1 6956526
## 2 doi 3 10.1016/0028-2243(82)90100-9
## 3 rid 8 6956526
## 4 eid 8 6956526
# 看被引用次数
taxize_summ$pmcrefcount
## [1] 2
可以接受多个ID
给entrez_summary()一个向量,给你返回一个列表
vivax_search <- entrez_search(db = "pubmed",
term = "(scRNA[TITL] AND 2019:2020[PDAT] )")
multi_summs <- entrez_summary(db="pubmed", id=vivax_search$ids)
# 多个ID结果的提取
extract_from_esummary(multi_summs, "fulljournalname")
# 提取多列
date_and_cite <- extract_from_esummary(multi_summs, c("pubdate", "pmcrefcount", "title"))
knitr::kable(tail(t(date_and_cite)), row.names=FALSE)
| pubdate | pmcrefcount | title |
|---|---|---|
| 2020 | Single-Cell Transcriptomics of Immune Cells: Cell Isolation and cDNA Library Generation for scRNA-Seq. | |
| 2020 Aug 20 | 10 | Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. |
| 2020 Dec 2 | 2 | A Distinct Transcriptional Program in Human CAR T Cells Bearing the 4-1BB Signaling Domain Revealed by scRNA-Seq. |
| 2020 Jul 10 | 1 | Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. |
| 2020 Jun 8 | Direct-seq: programmed gRNA scaffold for streamlined scRNA-seq in CRISPR screen. | |
| 2020 Jun | Detecting differential alternative splicing events in scRNA-seq with or without Unique Molecular Identifiers. |
4-ID完整信息(fetch)
entrez_summary提供的简略信息有时还不能满足要求,想看更完整的信息就用到了entrez_fetch,需要结合参数rettype来指定想要的具体内容
具体
rettype对应的关系看 :https://www.ncbi.nlm.nih.gov/books/NBK25499/table/chapter4.T._valid_values_of__retmode_and/
# 首先进行ID关联
gene_ids <- c(351, 11647)
linked_seq_ids <- entrez_link(dbfrom="gene", id=gene_ids, db="nuccore")
linked_transripts <- linked_seq_ids$links$gene_nuccore_refseqrna
head(linked_transripts)
## [1] "1907153913" "1889693417" "1869284273" "1676441520"
## [5] "1676319912" "1675178653"
# 然后拿到fasta序列
all_recs <- entrez_fetch(db="nuccore", id=linked_transripts, rettype="fasta")
class(all_recs)
## [1] "character"
nchar(all_recs)
## [1] 57671
# 可以不用完全打印出来,看看top lines
cat(strwrap(substr(all_recs, 1, 500)), sep="\n")
## >XM_006538498.4 PREDICTED: Mus musculus alkaline
## phosphatase, liver/bone/kidney (Alpl), transcript
## variant X2, mRNA
## AGGCCCTGTAACTCCTCCAAGAGAACACATGCCCAGTCCAGAGAAGAGCACAAGGTAGATCTTGTGACCA
## TCATCGGAACAAGCTGCAGTGGTAGCCTGGGTAGAAGCTGGCAGAGGGAGACCATCTGCAAACCAGGAAC
## GCTGTGAGAAGAGAAAGGACAGAGGTCCTGACATACTGTCACAGCCGCTCTGATGTATGGATCGGAACGT
## CAATTAACGTCAATTAACATCTGACGCTGCCCCCCCCCCCCTCTTCCCACCATCTGGGCTCCAGCGAGGG
## ACGAATCTCAGGGTACACCATGATCTCACCATTTTTAGTACTGGCCATCGGCACCTGCCTTACCAACTCT
## TTTGTGCCAGAGAAAGAGAGAGACCCCAG
还可以把这些序列输出
write(all_recs, file="my_transcripts.fasta")
返回XML格式数据
大部分的NCBI数据库都可以支持XML数据类型
Tt <- entrez_search(db="taxonomy", term="(Homo sapiens[ORGN]) AND Species[RANK]")
tax_rec <- entrez_fetch(db="taxonomy", id=Tt$ids, rettype="xml", parsed=TRUE)
class(tax_rec)
## [1] "XMLInternalDocument" "XMLAbstractDocument"
tax_list <- XML::xmlToList(tax_rec)
tax_list$Taxon$GeneticCode
## $GCId
## [1] "1"
##
## $GCName
## [1] "Standard"
# 还可以更复杂
tt_lineage <- tax_rec["//LineageEx/Taxon/ScientificName"]
tt_lineage[1:4]
## [[1]]
## <ScientificName>cellular organisms</ScientificName>
##
## [[2]]
## <ScientificName>Eukaryota</ScientificName>
##
## [[3]]
## <ScientificName>Opisthokonta</ScientificName>
##
## [[4]]
## <ScientificName>Metazoa</ScientificName>
XML::xpathSApply(tax_rec, "//LineageEx/Taxon/ScientificName", XML::xmlValue)
## [1] "cellular organisms" "Eukaryota"
## [3] "Opisthokonta" "Metazoa"
## [5] "Eumetazoa" "Bilateria"
## [7] "Deuterostomia" "Chordata"
## [9] "Craniata" "Vertebrata"
## [11] "Gnathostomata" "Teleostomi"
## [13] "Euteleostomi" "Sarcopterygii"
## [15] "Dipnotetrapodomorpha" "Tetrapoda"
## [17] "Amniota" "Mammalia"
## [19] "Theria" "Eutheria"
## [21] "Boreoeutheria" "Euarchontoglires"
## [23] "Primates" "Haplorrhini"
## [25] "Simiiformes" "Catarrhini"
## [27] "Hominoidea" "Hominidae"
## [29] "Homininae" "Homo"
5-练习
问题一:找到与某个基因有关联的其他基因
以ZNF365基因为例(zinc finger protein 365 [ Homo sapiens (human) ])【https://www.ncbi.nlm.nih.gov/gene/22891】
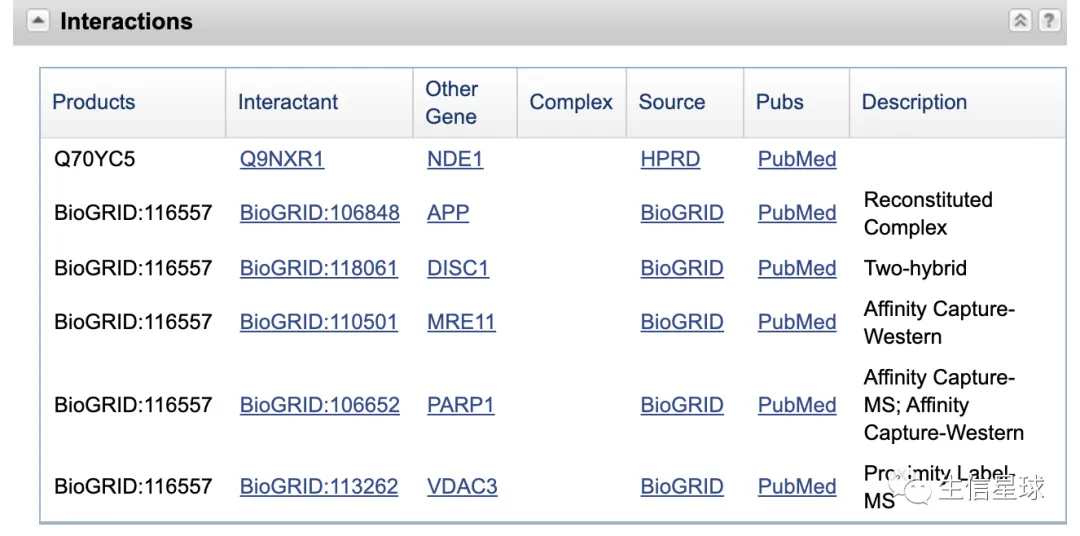
看看用代码怎么寻找
gene_search <- entrez_search(db="gene", term="(ZNF365[GENE]) AND (Homo sapiens[ORGN])")
gene_search
## Entrez search result with 1 hits (object contains 1 IDs and no web_history object)
## Search term (as translated): ZNF365[GENE] AND "Homo sapiens"[Organism]
interactions_from_gene <- function(gene_id){
xmlrec <- entrez_fetch(db="gene", id=gene_id, rettype="xml", parsed=TRUE)
XML::xpathSApply(xmlrec,
"//Gene-commentary[Gene-commentary_heading[./text()='Interactions']]//Other-source[Other-source_src/Dbtag/Dbtag_db[./text()='GeneID']]//Other-source_anchor",
XML::xmlValue)
}
interactions_from_gene(gene_search$ids)
## [1] "VDAC3" "MRE11" "PARP1" "APP" "DISC1" "NDE1"
问题二:根据核酸记录ID,提取相关文献
library(rentrez)
library(XML)
library(dplyr)
# function to add column to df if not already included
fncols <- function(data, cname) {
add <-cname[!cname%in%names(data)]
if(length(add)!=0) data[add] <- NA
data
}
# function to collate all publications associated with sequences
get_pub_info <- function(i){
fetch2 <- entrez_fetch(db = "nucleotide", id = i,
rettype = "gbc", retmode="xml", parsed = TRUE)
xml_list2 <- xmlToList(fetch2)
ref_list <- xml_list2$INSDSeq$INSDSeq_references
# extract publication fields info
authors <- unlist(ref_list$INSDReference$INSDReference_authors) %>% paste(collapse = "; ")
title <- ref_list$INSDReference$INSDReference_title
journal <- ref_list$INSDReference$INSDReference_journal
year <-gsub(".*\\((.*)\\).*", "\\1", journal)
pm_id <- ref_list$INSDReference$INSDReference_pubmed
remark <- ref_list$INSDReference$INSDReference_remark
# create data frame of information
pub.data <- data.frame(i, authors, journal, year)
if(is.null(title)==FALSE) pub.data$title <- title
if(is.null(pm_id)==FALSE) pub.data$pubmed_id <- pm_id
if(is.null(remark)==FALSE) pub.data$remark <- remark
pub.data <- fncols(pub.data, c("title", "pubmed_id", "remark"))
}
sequence_list <- c("AB687721.2", "AB600942.1", "AJ880277.1")
list_of_dfs <- lapply(sequence_list, get_pub_info) # run function on list of sequences
df_combine <- bind_rows(list_of_dfs)
colnames(df_combine)[1] <- "NCBI_idv"
df_combine <- tidyr::separate(df_combine, remark, c("text", "doi"), sep = "DOI:") # extract doi
df_combine <- tidyr::separate(df_combine, doi, c("doi", "text2"), sep = ";")
df_combine$remark <- paste(df_combine$text,df_combine$text2)
df_combine$text <- NULL
df_combine$text2 <- NULL
head(df_combine[, c("pubmed_id", "doi", "journal")])
## pubmed_id doi
## 1 23135729 10.1128/JVI.02419-12
## 2 8709862 <NA>
## 3 16525735 <NA>
## journal
## 1 J. Virol. 87 (2), 1105-1114 (2013)
## 2 Microbiol Immunol 40 (4), 271-275 (1996)
## 3 Virus Genes 32 (1), 49-57 (2006)