224-ChIP-Seq如果有多个生物重复应该怎么评价?
刘小泽写于2021.1.6 内容来自:https://hbctraining.github.io/Intro-to-ChIPseq/lessons/07_handling-replicates-idr.html
前言
和任何的高通量测序一样,一个样本只设置一个重复会出现假阳性的问题,以转录组为例,现在最少要求设置3个生物重复。当然,ChIP-Seq的实验操作难度比常规的RNA-Seq要大,而且步骤繁琐、限制因素也比较多,所以现在很多都不设置重复。但这不代表生物重复就不必要,很多高质量的研究成果(包括ENCODE的官方推荐)都是设置了重复。这里假设每个处理做了两个生物重复,那么来看看如何对组内重复进行评价吧。
比较简单的做法
对每个处理所有的重复样本分别进行call peaks,然后用bedtools取交集,从而得知实验重复做的好不好。
这个操作可以参考:https://hbctraining.github.io/Intro-to-ChIPseq/lessons/handling-replicates-bedtools.html

bedtools intersect \
-a macs2/Nanog-rep1_peaks.narrowPeak \
-b macs2/Nanog-rep2_peaks.narrowPeak \
-wo > bedtools/Nanog-overlaps.bed
看一下这个-wo参数的作用:
# When an interval in A does not intersect an interval in B, it will not be reported
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
这个方案需要思考的是:既然我们需要基于peaks结果,那么在call peaks过程中,设置的阈值会不会对后续有影响?另外,我们要取交集,那么bedtools对于交集的定义是不是也是人为定义的?到底是1bp就说有交集还是5bp才算交集呢?
使用IDR方法
是什么?
IDR全称是:Irreproducibility Discovery Rate(不可重复率)。它可以根据多个生物重复的peaks结果提取一致性最高的peak区间。代码在:https://github.com/nboley/idr
在判断真实信号值与噪音方面,它和spearman相关性方法不一样,不需要人工设置阈值来判断,软件会拿原始的peaks文件,自动根据peaks信号值在重复样本的分布来确定合适的阈值,进而判断真实信号值
Unlike the usual scalar measures of reproducibility, the IDR approach creates a curve, which quantitatively assesses when the findings are no longer consistent across replicates.
有了真实信号值以后,会对peaks的信号进行排序(可以根据pvalue、qvalue或者fold enrichment),然后每个信号值会有一个IDR value,用来表示该信号在重复样本中的一致性,数值越大越难重复。因此最后只需要找到低于设置的IDR阈值的peaks信号
为什么?
- 可以直接使用最原始的call peaks结果,而不需要人工预先设置阈值进行筛选
- 对数据分布不进行任何先验假设,只需要将信号值根据fold enrichment、pvalue或者qvalue进行排序
- ENCODE和modENCODE计划广泛使用IDR方法进行生物重复peaks的评价
怎么做?
安装
# conda
conda install -c bioconda idr
# Ubuntu 14.04+
(sudo) apt-get install python3-dev python3-numpy python3-setuptools python3-matplotlib
首先是call peaks
# Call peaks using a liberal p-value cutoff
macs2 callpeak -t treatFile.bam -c inputFile.bam -f BAM -g 1.3e+8 -n macs/NAME_FOR_OUPUT -B -p 1e-3 2> macs/NAME_FOR_OUTPUT_macs2.log
当然,IDR可以与其他的peaks caller搭配,例如:
- SPP - Works out of the box
- MACS1.4 - DO NOT use with IDR
- MACS2 - Works well with IDR with occasional problems of too many ties in ranks for low quality ChIP-seq data.
- HOMER - developers have a detailed pipeline and code (in beta) for IDR analysis with HOMER at https://github.com/karmel/homer-idr
- PeakSeq - Run with modified PeakSeq parameters to obtain large number of peaks
- HotSpot, MOSAiCS, GPS/GEM, …
然后是排序
#Sort peak by -log10(p-value)
sort -k8,8nr NAME_OF_INPUT_peaks.narrowPeak > macs/NAME_FOR_OUPUT_peaks.narrowPeak
再比较两个重复
idr --samples Nanog_Rep1_sorted_peaks.narrowPeak Nanog_Rep2_sorted_peaks.narrowPeak \
--input-file-type narrowPeak \
--rank p.value \
--output-file Nanog-idr \
--plot \
--log-output-file nanog.idr.log
得到的结果 Nanog-idr 中前10列是标准的narrowPeak文件,需要注意是第5列,它是scaled IDR value,计算方法是:min(int(-125*log2(IDR), 1000)。
- 比如一个peak的IDR是0(重复性非常好),那么对应的scaled IDR value就是
min(int(-125*log2(0), 1000)=1000; - 如果IDR是0.05,那么scaled IDR value就是
min(int(-125*log2(0.05), 1000)=540; - 如果IDR是1(重复性非常差),那么scaled IDR value就是
min(int(-125*log2(1.0), 1000)=0
这时可以统计一下有多少区间的IDR<0.05
awk '{if($5 >= 540) print $0}' Nanog-idr | wc -l
除此以外,第11列是:-log10(local IDR value),第12列是:-log10(global IDR value)
第15列是:ranking measure from Rep1 ,第19列是ranking measure from Rep2
最后
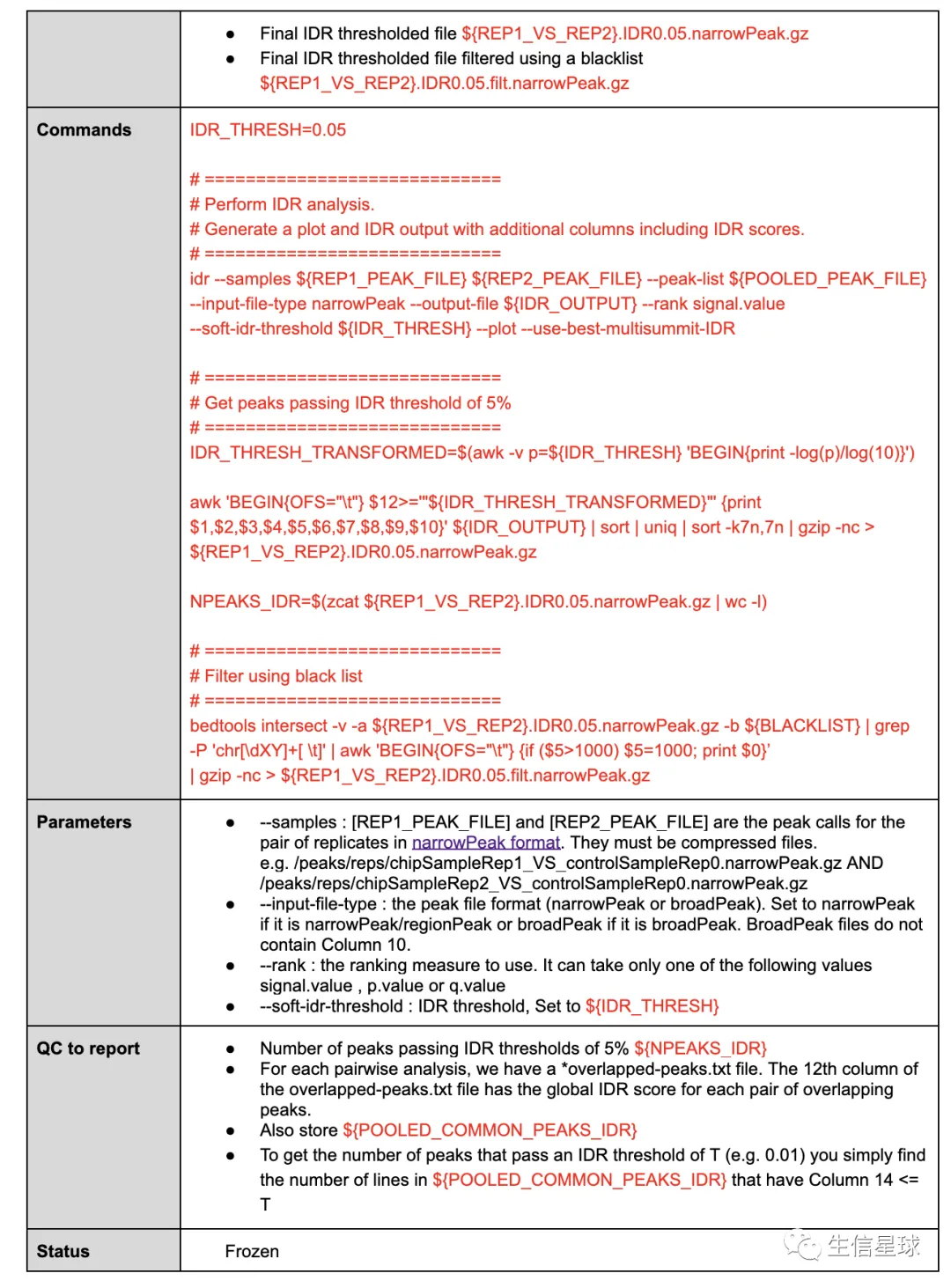
ENCODE的ChIP-Seq分析pipeline也可以参考:
https://docs.google.com/document/d/1lG_Rd7fnYgRpSIqrIfuVlAz2dW1VaSQThzk836Db99c/edit
其中也记录了IDR的使用: