216-为什么你的热图画出来是“糊”的?
刘小泽写于2020.11.30
前言
最近在分析一个单细胞数据时,使用seurat去做marker基因的热图,发现热图竟然是糊的,cluster之间的marker竟然没有什么差别。
看看具体原因出在哪
正确示例:以PBMC 3k数据为例
原始数据在:https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
可以下载并加载
pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
然后我这里已经做好了RData (https://share.weiyun.com/9HZRMIWe),直接加载即可
load(file = 'pbmc.Rdata')
> pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
快速走一下标准流程(详细流程可以看:https://www.jieandze1314.com/post/cnposts/scrna-19/)
# 质控
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
# 过滤
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
> pbmc
An object of class Seurat
13714 features across 2638 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
# 归一化
pbmc <- NormalizeData(pbmc)
# 找变化比较大的基因
pbmc <- FindVariableFeatures(pbmc,
selection.method = "vst", nfeatures = 2000)
如果此时直接进行PCA,会给出错误提示,让你先进行scale操作
pbmc <- RunPCA(object = pbmc, pc.genes = VariableFeatures(pbmc))
# Error in PrepDR(object = object, features = features, verbose = verbose) :
# Data has not been scaled. Please run ScaleData and retry
因此,在FindVariableFeatures()之后,一般要先进行scale操作
pbmc <- ScaleData(pbmc)
pbmc <- RunPCA(object = pbmc, pc.genes = VariableFeatures(pbmc))
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
table(pbmc@meta.data$RNA_snn_res.0.5)
# 0 1 2 3 4 5 6 7 8
# 697 483 480 344 271 162 155 32 14
set.seed(123)
pbmc <- RunTSNE(object = pbmc, dims = 1:10, do.fast = TRUE)
pbmc <- RunUMAP(pbmc, reduction = "pca", dims = 1:10)
sce.markers <- FindAllMarkers(object = pbmc, only.pos = TRUE, min.pct = 0.25, thresh.use = 0.25)
library(dplyr)
top10 <- sce.markers %>% group_by(cluster) %>% top_n(10, avg_logFC)
DoHeatmap(pbmc,top10$gene,size=3)

上面的图基本是正常的情况,能够看出每个cluster的差异
再来一个错误示例
例如这样的图,你会看到虽然得到了marker gene,但在图中表现却很差,很多都看不出差别
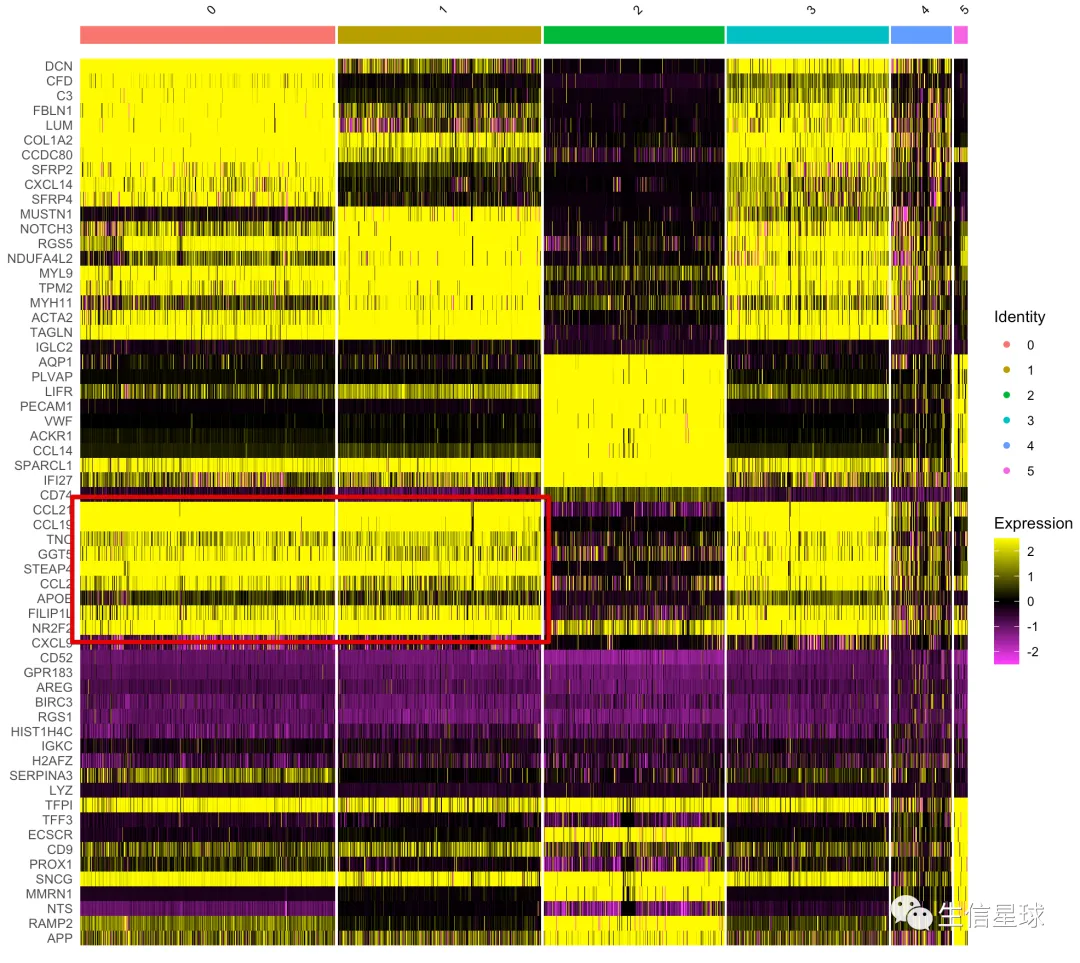
Tip1:当你看到类似上图,就要意识到:
你的数据可能没有进行scale操作,还记得它是干啥的吗?
NormalizeData进行的归一化主要考虑了测序深度/文库大小的影响(reads*10000 divide by total reads, and then log),但实际上归一化的结果还是存在基因表达量分布区间不同的问题; 而ScaleData做的就是在归一化的基础上再添zero-centres(mean/sd =》 z-score)
也就是做了两件事:
- 将每个基因在所有细胞的表达量进行调整,使得每个基因在所有细胞的表达量均值为0
- 将每个基因的表达量进行标准化,让每个基因在所有细胞中的表达量方差为1
是不是想到了我们用pheatmap画普通热图的时候,也会加上pheatmap(gp_expr, scale = "row") 这样的操作,才能尽可能减少极值的影响
如果重新加上scale的操作后,就变成了正常的样子:

Tip2:之前不是说不进行scale就没办法继续进行PCA吗?那为什么还会出现上面忘记scale但依然运行的情况?
是的,PCA是依赖于scale的结果的,但是如果你的数据不是单一的,而是经过了多个数据集的整合,一般会先整体进行一个scale操作。不过后面自己还是会继续分亚群,而且每个亚群会继续分析。那么这时每个亚群的数据即使不单独进行scale,也是可以运行PCA的。
Tip3: 是不是必须每个亚群分析都要重新scale呢?
这个问题存在争议
来自:https://github.com/satijalab/seurat/issues/3144
支持重新找高变化基因,重新scale的:
- re-find highly variable genes before scaling data on your subset dataset

反对意见:
- just use the scaled data in the original database to perform PCA in subset dataset without rescaling

来自:https://github.com/satijalab/seurat/issues/1883
引用一篇Cell paper的方法:https://doi.org/10.1016/j.cell.2019.06.029
subset() the population of interest and run the complete preprocessing only on those cells again
有人提出:
Choose a subset of cells, and use the integration assay to Run PCA, umap, findneighbors and findclusters to do subclustering.
当然还有官方的回答:https://github.com/satijalab/seurat/issues/752

官方还在https://github.com/satijalab/seurat/issues/1347中提到了:
you could rescale after doing a subset on your data
另外,也有人搬出了自己使用的脚本(数据整合+亚群重分析):
commongenes_tokeep <- Reduce(intersect, lapply(SCT_Integrated.anchors@object.list, rownames))
SCT_integrated <- IntegrateData(anchorset = SCT_Integrated.anchors, normalization.method = "SCT", features.to.integrate = commongenes_tokeep)
SCT_integrated <- RunPCA(SCT_integrated)
SCT_integrated <- RunUMAP(SCT_integrated, dims = 1:30)
SCT_integrated <- FindNeighbors(SCT_integrated, dims = 1:30)
SCT_integrated <- FindClusters(SCT_integrated, resolution=0.8)
DefaultAssay(Sample_subset) <- "RNA"
Sample_subset <- subset(SCT_integrated, idents = c(0, 2, 3, 7))
Sample_subset <- ScaleData(Sample_subset, vars.to.regress = c("percent.mt")
Sample_subset <- RunPCA(Sample_subset)
Sample_subset <- RunUMAP(Sample_subset, dims = 1:30)
Sample_subset <- FindNeighbors(Sample_subset, dims = 1:30)
Sample_subset <- FindClusters(Sample_subset, resolution = 0.8)