215-ChIP-Seq的blacklist黑名单
刘小泽写于2020.3.25 偶然看到有的文章中写到了ChIP-Seq分析中,在call peaks之前特意去除了blacklist,这个“黑名单”是啥?来探索一下
什么是blacklist?
可以参考:Carroll et al. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3989762/
基因组上会存在一些特定的重复序列,例如在丝粒、端粒以及卫星重复序列,特点就是重复序列区域的碱基完全相同。而二代测序的数据进行比对时(比如现在有重复区域A和B,而A和B的碱基完全相同),仅仅依靠比对的算法,是不能判断reads比对到A还是B的。这时不同的软件会进行判断:有的软件会随机选取一个,有的软件会两个区域都进行计算。这种计算上的不确定性导致了这些区域的测序深度普遍偏高。
回归ChIP-Seq数据,我们一般是通过比较IP组和input组之间基因组上测序深度的差异,通过这个差异再加上一些计算方法,macs2等软件就会帮我们得到一系列peaks,也就得到了结合位点附近区域。如果我们是通过测序深度来确定peaks,那么重复序列区域的虚高情况势必会造成影响。因此,这部分区域被列入了“黑名单”。
为啥考虑blacklist?
参考:https://www.biostars.org/p/361297/
It’s still considered best-practice to remove these regions. For genomes like GRCh38, the blacklisted regions are largely comprised of things like major satellite repeats, which are primarily located in hard-masked telomeric and pericentromeric regions.
参考:https://deeptools.readthedocs.io/en/develop/content/feature/blacklist.html
引入假阳性peaks,对样本文库归一化有影响(Including these regions can lead not only to false-positive peaks, but can also throw off between-sample normalization.)
因此,去除blacklist region主要就是为了降低peaks的假阳性
怎么去除blacklist?
Devon Ryan在https://bioinformatics.stackexchange.com/questions/458/when-to-account-for-the-blacklisted-genomic-regions-in-chip-seq-data-analyses/459#459?newreg=dca76bad61c443d7b4f0b1abd1487878中提到: 如果仅仅要去掉这些区域是很简单的;一般在peak calling之前去除这些区域;这个去除对peak calling结果的改进不大;deeptools可以设置blacklist region;基因组版本越新(像GRCh38 and GRCm38), blacklisted regions范围就越小,所以现在很多时候也不用考虑这些blacklist区域
另外 Devon在https://www.biostars.org/p/238222/中提到,基因组是不断完善的:
Only 10% of the problematic regions in hg18 were still problematic in hg19. Somewhere Heng Li has a presentation showing further improvements in GRCh38. This is the same for the mm8 -> mm9 -> mm10 progression of releases. 另外,不用纠结一开始就去除相关的reads,那样也比较费时;我们使用的QC软件都是考虑到这些区域的;只需要在bed文件中去掉这些就好了,也方便操作
首先获得blacklist列表
如果要自己创建,要使用的计算资源和时间可以参考:https://github.com/Boyle-Lab/Blacklist RAM: 64+ GB CPU: 24+ cores, 3.4+ GHz/core Approximately 192 CPU hours
参考一:
http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/

参考二:另外ENCODE也提供了blacklist:https://www.encodeproject.org/search/?type=Annotation&annotation_type=blacklist
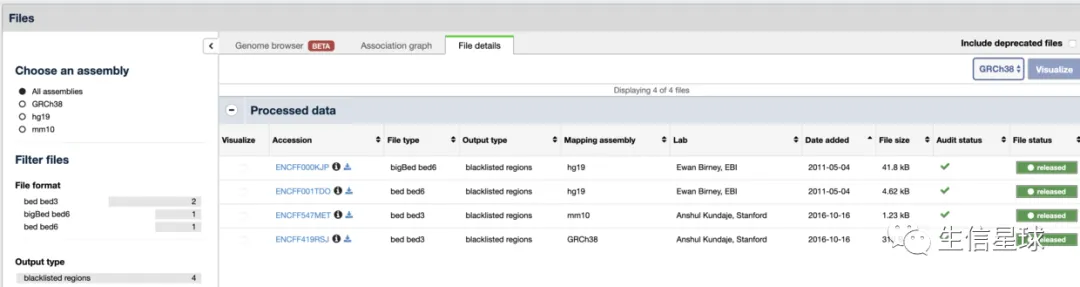
参考三:2019年又有人更新了blacklist:https://github.com/Boyle-Lab/Blacklist/tree/master/lists 还发了篇文章
These blacklists are described in The ENCODE Blacklist: Identification of Problematic Regions of the Genome (June 2019):
Here, we define the ENCODE blacklist- a comprehensive set of regions in the human, mouse, worm, and fly genomes that have anomalous, unstructured, or high signal in next-generation sequencing experiments independent of cell line or experiment.
参考四:https://sites.google.com/site/anshulkundaje/projects/blacklists
参考五:【应该也是最新版】https://sites.google.com/site/anshulkundaje/projects/blacklists
- NEW: VERSION 3 (05/20/2020)
- HUMAN (hg38/GRCh38): https://www.encodeproject.org/files/ENCFF356LFX/ (Manually curated)
- HUMAN (hg19/GRCh37): ENCODE portal link: https://www.encodeproject.org/files/ENCFF001TDO/ (Manually curated, same as version 1)
- For other species you can use the Version 2 blacklists at https://github.com/Boyle-Lab/Blacklist/tree/master/lists
然后最简单的办法:
bedtools intersect -v -a your_regions.bed -b blacklist.bed
# -v:Only report those entries in A that have no overlap in B
另外,deeptools可以设置blacklist region
https://deeptools.readthedocs.io/en/develop/content/feature/blacklist.html
deepTools全程支持指定–blackListFileName参数
小Tip
如果遇到blacklist文件出现overlaps问题
参考:https://www.biostars.org/p/171354/
如果从http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/这里下载,可能使用的blacklist文件中会有重叠,例如会存在这样的区域:
chr16 34586660 34587100
chr16 34587060 34587660
使用:bedtools merge -i blacklist.bed 把有重叠的区域合并起来
除此以外,还有SV blacklist
10x genomics has a topic about 【SV Calling Filter File】: https://support.10xgenomics.com/genome-exome/software/pipelines/latest/advanced/references
# The filter file is used by the SV algorithm to reduce false positives due to gaps in the reference, known or putative assembly issues such as unplaced contigs, and highly polymorphic regions.
wget http://cf.10xgenomics.com/supp/genome/hg19/sv_blacklist.bed
wget http://cf.10xgenomics.com/supp/genome/b37/sv_blacklist.bed
wget http://cf.10xgenomics.com/supp/genome/GRCh38/sv_blacklist.bed
如果要去除线粒体的影响
在ATAC-Seq中,会有这种需求:https://www.biostars.org/p/178574/
准备两个文件:
- ATAC_seq author mitochondrial blacklist(https://sites.google.com/site/atacseqpublic/atac-seq-analysis-methods/mitochondrialblacklists-1)
- ENCODE signal artifact blacklist(http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse/)
再来个批量操作
for i in *.bedfile; do bedtools intersect -v -a $i -b [PATH]/mitochondrial.blacklist.bed [PATH]/signal.artifact.blacklist.bed > $i.bed; done
如果是从bam中要去除线粒体的reads
这个比bed操作要复杂一点,但思路简单:首先要把比对后的bam文件构建索引,然后从头文件中把chrM的去掉即可
for i in *.bam; do non_chrM_list=$(samtools view -H $i | grep chr | cut -f2 | sed 's/SN://g' | grep -v chrM) samtools view -b $i $non_chrM_list -o [outfilename.bam]; done;
# 检查是否去除了chrM reads
grep chrM [outfilename.bam]