208-详细回顾非模式物种注释构建过程
刘小泽写于2020.10.12 有日子没写推送了,这段时间一直在忙其他的事情 最近被问到如何对非模式生物进行富集分析,其实之前我硕士就是做非模式生物的,所以当时探索的比较多。但后来转做人类数据,所以慢慢接触就少了。 之前写的在:https://www.jieandze1314.com/post/cnposts/78/ 这次也算是对之前的一个总结,将之前的代码做了更详细的注释以及更新
富集分析的核心是什么?
我们拿到一些基因列表,想知道它们在什么通路比较多。先不讨论其中的统计知识,但也应该知道我们首先要得到各个通路吧,然后每个通路又有一定数量的基因吧。那么我们的这些基因有多少落在其中呢?
因此,第一个关键点就是知道这个物种有多少个通路以及每个通路中的基因;
第二个关键就是我们落在其中的数量,是不是能达到统计学上的显著性
如果是模式生物或者常见的物种是最简单的
这些研究比较广泛的物种在:http://bioconductor.org/packages/release/BiocViews.html#___OrgDb 可以自己查询
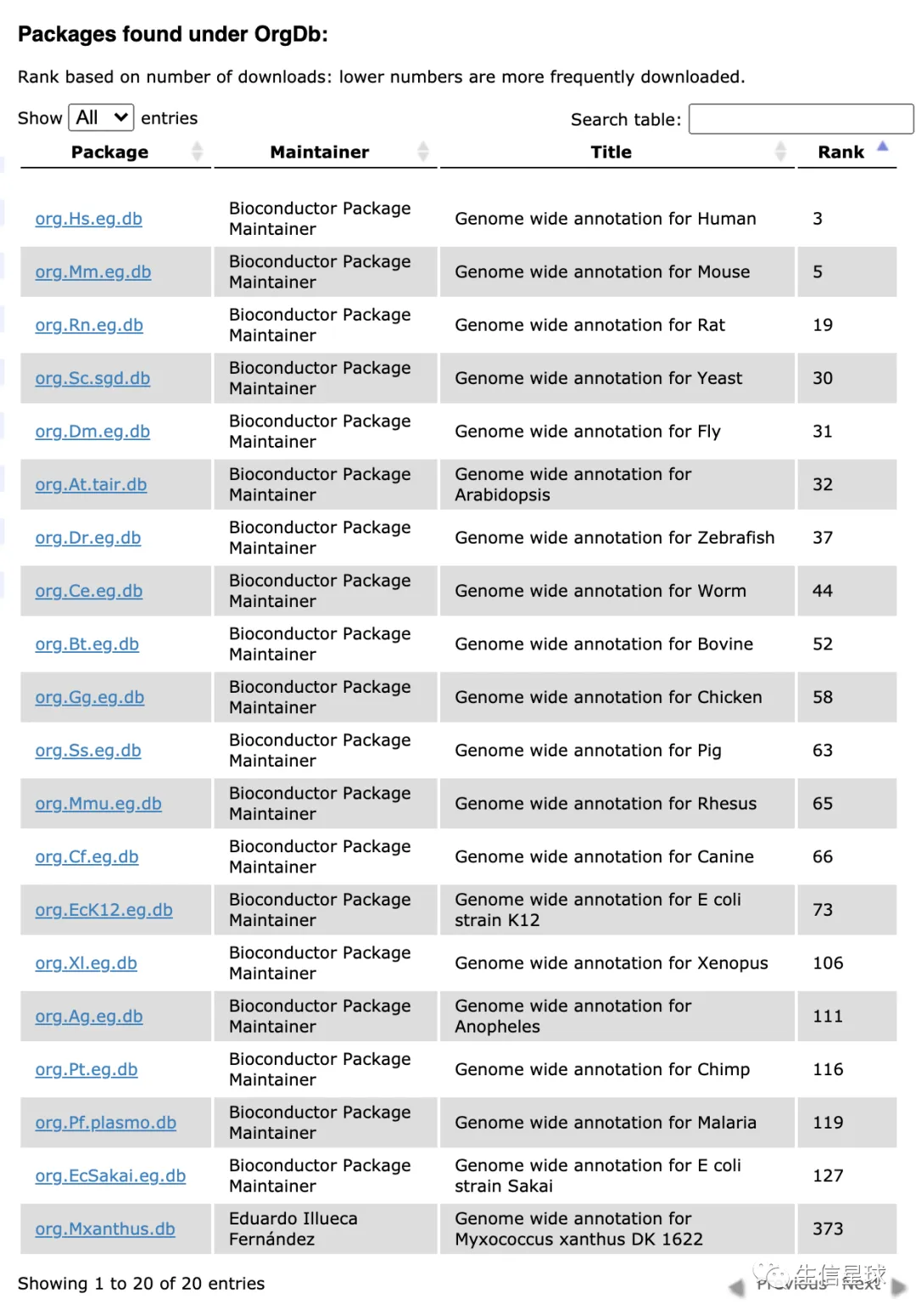
看到有org.xxx.db了,其实可以暗自庆幸,因为有非常成熟的代码供参考
# 比如对人类下调基因富集
BP_down <- enrichGO(gene = gs_dn,
keyType = 'ENSEMBL',
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.01,
readable = TRUE)
如果没有现成的OrgDb,还有两个方法
方法一:看看AnnotationHub有没有
这个是次优方案,因为有的物种虽然没有研究的特别多,但毕竟也是有人为它做了注释的(比如我之前研究的棉铃虫)
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
options("repos" = c(CRAN="http://mirrors.cloud.tencent.com/CRAN/"))
options(download.file.method = 'libcurl')
options(url.method='libcurl')
if (!requireNamespace("BiocManager", quietly = TRUE)
install.packages("BiocManager"))
BiocManager::install("AnnotationHub", version = "3.8")
library(AnnotationHub)
然后加载AnnotationHub其中的物种注释结果
hub <- AnnotationHub()
# 查看方法一:调用图形界面查看物种
display(hub)
# 查看方法二:根据物种拉丁文名称查找
query(hub,"helicoverpa")
# 如果有返回结果的话,可以下载sqlite数据库
ha.db <- hub[['AH66950']]
#查看前几个基因(Entrez命名)
head(keys(ha.db))
#查看包含的基因数
length(keys(ha.db))
#查看包含多少种ID
columns(ha.db)
#查看前几个基因的ID
select(ha.db, keys(ha.db)[1:3],
c("REFSEQ", "SYMBOL"), #想获取的ID
"ENTREZID")
如果返回的结果为空,那么继续探索下一个方法
方法二:利用AnnotationForge自行构建
需要基于eggnog的注释结果进行操作,它相当于一个“桥梁”,连接基因与GO、KEGG等通路
eggnog的重点就是: 1:将自己的核酸序列与Eggnog中的蛋白序列进行比对(eggnog借助了blastp)【这个过程可以是基于相似性(DIAMOND),也可以是基于结构域(HMMER)】=》 2:比对完就知道我们的序列与哪个物种的哪个直系同源组(Orthologous Group)最相近=》 3:利用各个数据库的关联进行COG、GO、Pathway的功能分类
如果使用在线版:http://eggnog-mapper.embl.de/
如果使用本地版:首先安装eggnog
这个软件基于python2.7,所以需要在python2.7的小环境中进行安装
conda install -c bioconda eggnog-mapper -y
如果使用本地版:下载数据库
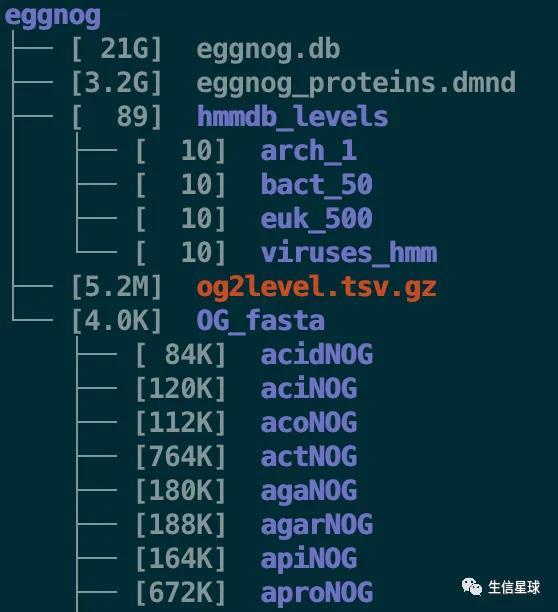
下载真核、细菌、古细菌、病毒(euk、bact、arch、viruses)的数据库
需要注意:需要200G左右的硬盘空间,并且对于真核生物需要90GB的内存,细菌大约32G内存,古细菌10G内存
download_eggnog_data.py euk bact arch viruses -y --data_dir eggnog
如果使用本地版:运行
emapper.py -m diamond -i test.fa -o diamond
有了运行结果以后,就到了R语言的部分
0 先对eggnog的原始结果稍微做点修改
主要是在shell中去掉开头的#,为了保证后面读取的顺利
sed '/^# /d' raw_eggnog_annotations.tsv | sed 's/#//' > emapper.annotations.tsv
1 读取数据
rm(list = ls())
options(stringsAsFactors = F)
library(tidyverse)
library(AnnotationForge)
# 当你发现用read.table, read.csv出错时,用import是个不错的选择
egg <- rio::import('emapper.annotations.tsv')
egg[egg==""] <- NA
> colnames(egg)
# [1] "#query_name" "seed_eggNOG_ortholog" "seed_ortholog_evalue" "seed_ortholog_score"
# [5] "best_tax_level" "Preferred_name" "GOs" "EC"
# [9] "KEGG_ko" "KEGG_Pathway" "KEGG_Module" "KEGG_Reaction"
# [13] "KEGG_rclass" "BRITE" "KEGG_TC" "CAZy"
# [17] "BiGG_Reaction" "V18" "V19" "V20"
# [21] "V21" "V22"
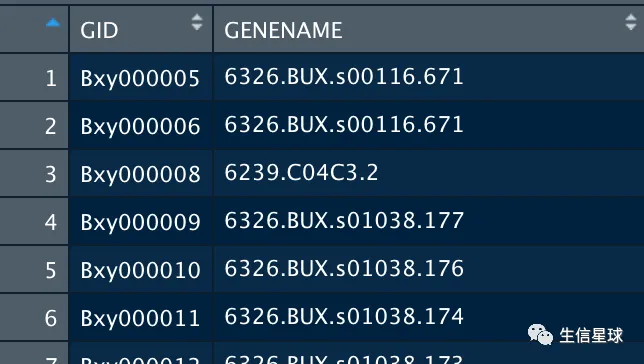
2 提取基因ID
重点是需要两列,一列是ID,一列是name(第二列不一定是真的name,但必须要有,比如看下面的示例)
# 要注意,要保证你的数据中存在query_name和seed_eggNOG_ortholog这样的列名下面代码才能运行,或者修改代码匹配自己的列名即可【之后的代码同理】
gene_info <- egg %>%
dplyr::select(GID = query_name, GENENAME = seed_eggNOG_ortholog) %>% na.omit()
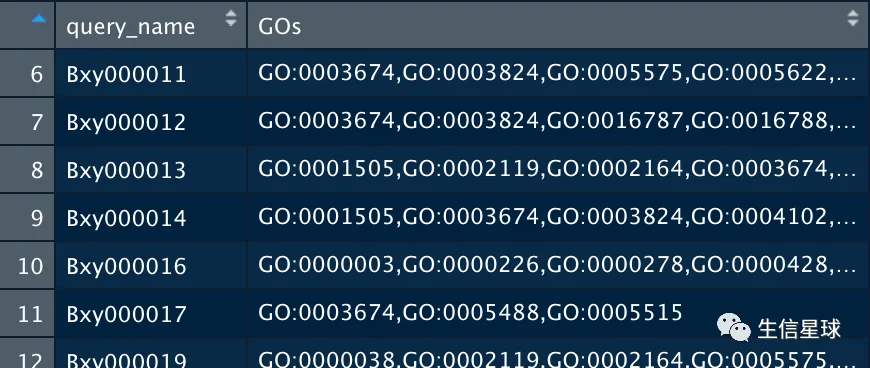
3 提取基因与GO的对应
gterms <- egg %>%
dplyr::select(query_name, GOs) %>% na.omit()
4 再将gterms的每一行中GO按照逗号分开
将实现的效果是:
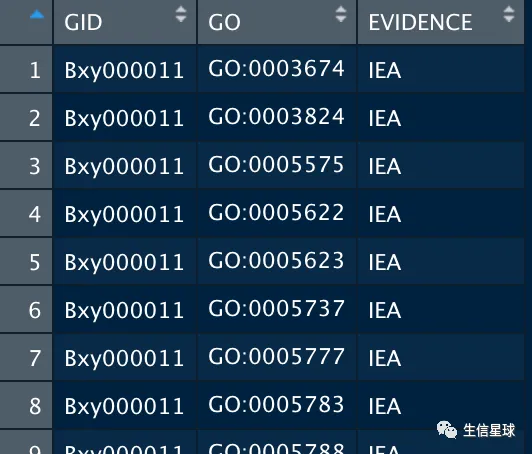
代码是:
# 之前的代码是用for循环,但是速度很感人,现在直接上sapply
library(stringr)
all_go_list=str_split(gterms$GOs,",")
gene2go <- data.frame(GID = rep(gterms$query_name,
times = sapply(all_go_list, length)),
GO = unlist(all_go_list),
EVIDENCE = "IEA")
5 提取基因与KEGG的对应
gene2ko <- egg %>%
dplyr::select(GID = query_name, KO = KEGG_ko) %>%
na.omit()

6 对json文件操作
https://www.genome.jp/kegg-bin/get_htext?ko00001
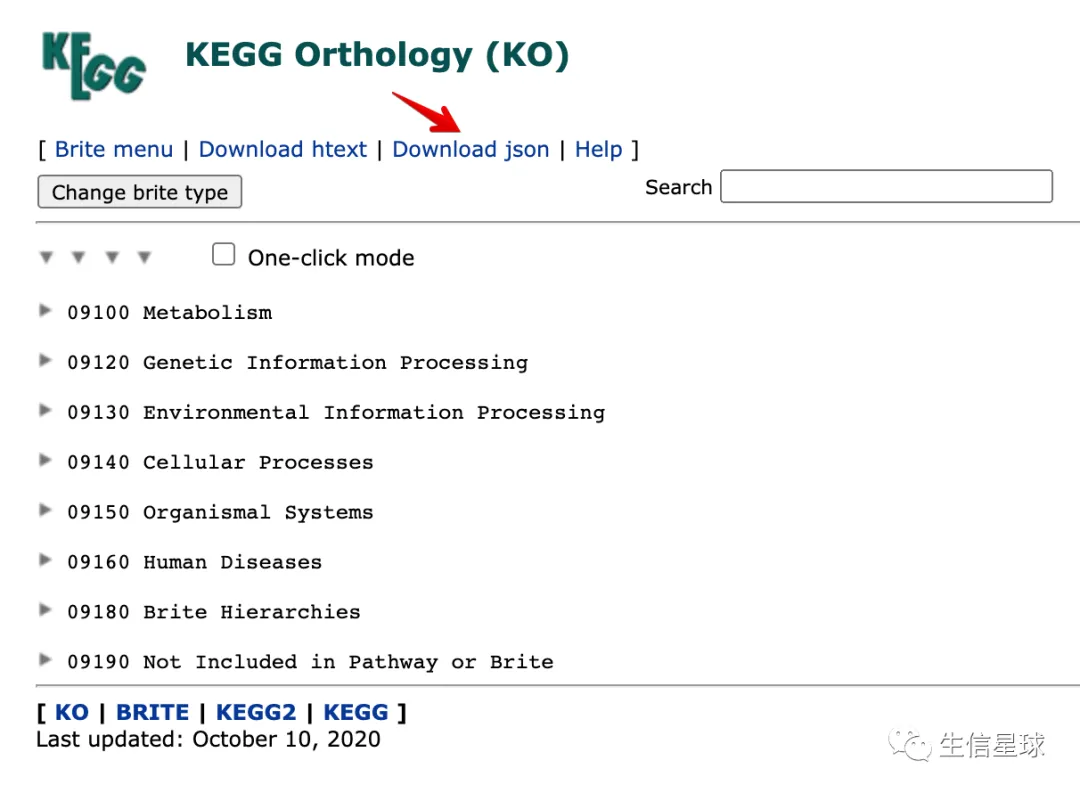
下载下来以后,就是一顿操作猛如虎,也没有必要了解代码每步是啥意思。中心思想就是:构建了一个函数,利用KEGG的json来获取ko与通路以及K与ko的对应关系
if(!file.exists('kegg_info.RData')){
library(jsonlite)
library(purrr)
library(RCurl)
update_kegg <- function(json = "ko00001.json",file=NULL) {
pathway2name <- tibble(Pathway = character(), Name = character())
ko2pathway <- tibble(Ko = character(), Pathway = character())
kegg <- fromJSON(json)
for (a in seq_along(kegg[["children"]][["children"]])) {
A <- kegg[["children"]][["name"]][[a]]
for (b in seq_along(kegg[["children"]][["children"]][[a]][["children"]])) {
B <- kegg[["children"]][["children"]][[a]][["name"]][[b]]
for (c in seq_along(kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]])) {
pathway_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["name"]][[c]]
pathway_id <- str_match(pathway_info, "ko[0-9]{5}")[1]
pathway_name <- str_replace(pathway_info, " \\[PATH:ko[0-9]{5}\\]", "") %>% str_replace("[0-9]{5} ", "")
pathway2name <- rbind(pathway2name, tibble(Pathway = pathway_id, Name = pathway_name))
kos_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]][[c]][["name"]]
kos <- str_match(kos_info, "K[0-9]*")[,1]
ko2pathway <- rbind(ko2pathway, tibble(Ko = kos, Pathway = rep(pathway_id, length(kos))))
}
}
}
save(pathway2name, ko2pathway, file = file)
}
update_kegg(json = "ko00001.json",file="kegg_info.RData")
}

你可能会好奇,这个K、ko有啥区别呢? 这个我之前也介绍过:https://www.jieandze1314.com/post/cnposts/185/ 因此,就更加明白了我们为什么要利用eggnog寻找直系同源组了:转录本对应到K(大写),然后K(大写)对应到ko(图中就明白它代表通路信息),最后就得到了通路名称

7 利用gene2ko与ko2pathway将基因与pathway对应起来
# 在运行数据框合并前,需要做到两个数据框的列名是对应的,并且将原来gene2ko中的ko修改一下
colnames(ko2pathway)=c("KO",'Pathway')
library(stringr)
gene2ko$KO=str_replace(gene2ko$KO,"ko:","")
# 合并代码是:
gene2pathway <- gene2ko %>% left_join(ko2pathway, by = "KO") %>%
dplyr::select(GID, Pathway) %>%
na.omit()
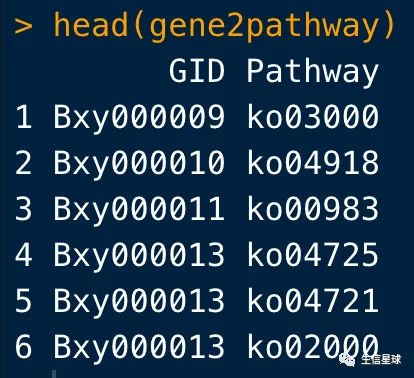
8 现在有了基因分别于GO、KEGG的对应,下面就要构建了
注意:
author和maintainer是需要按这个格式来写的,否则会在后面报错
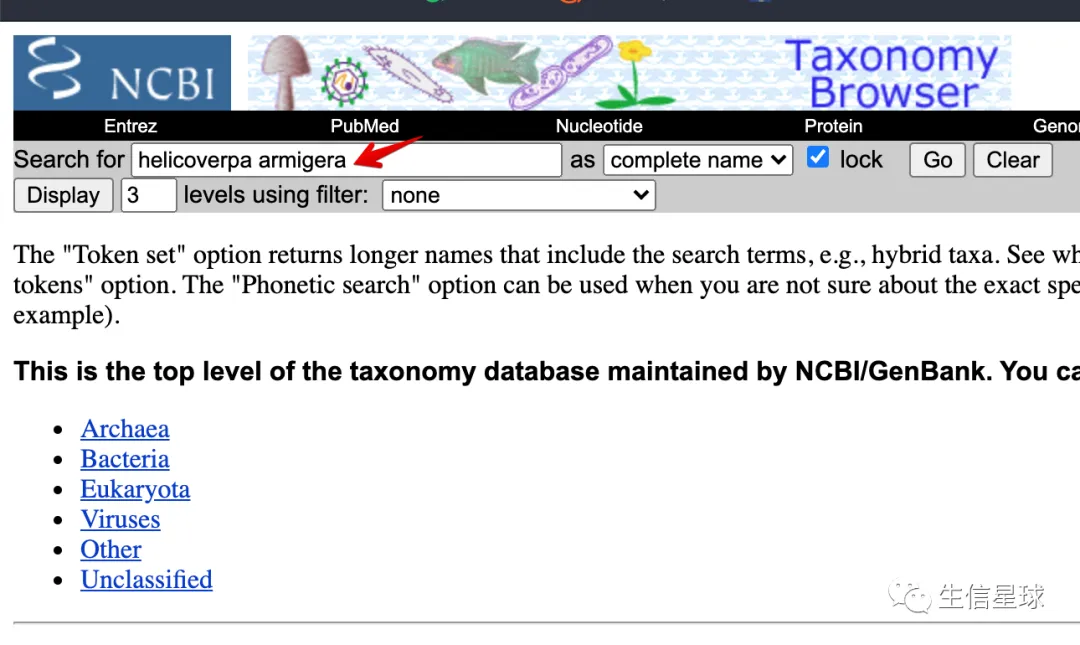
物种的
tax_id是可以输入物种的拉丁名查到的:https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi
library(AnnotationForge)
makeOrgPackage(gene_info=gene_info,
go=gene2go,
ko=gene2ko,
maintainer='doudou <yourmail@mail.com>',
author='doudou <yourmail@mail.com>',
pathway=gene2pathway,
version="0.0.1",
outputDir = ".",
tax_id=tax_id,
genus=genus,
species=species,
goTable="go")
这些信息都填好后,运行即可,会构建出一个org.xxx.eg.db的文件夹
9 像安装R包一样,来安装物种注释包
install.packages('org.your_species.eg.db',repos = NULL, type="source")
# 然后加载
library(org.your_species.eg.db)
10 最后,就可以随意富集分析啦!
注意:下面的
keyType要用GID,如果前面的列名和我保持一样的话,否则就改成自己包含GID的那个列名即可
ego_all <- enrichGO(gene = glist,
keyType="GID",
#模式物种
#OrgDb = org.Mm.eg.db,
#非模式物种
OrgDb = org.your_species.eg.db,
ont = "ALL", #ALL或BP或MF或CC
pAdjustMethod = "BH",
#pvalueCutoff = 0.01,
qvalueCutoff = 0.05)
写在最后
这次的版本,应该是比较完整的了,趁着这次机会也算实现了长期以来的一个小愿望吧。
每次操作这个物种注释过程,都会想起过去的初学时光,那时的自己真的是“痛并快乐着”,但好在有花花一直在💏(这个戏份不知道花花能不能看到),可以相扶相持,未来可期