203-生信星球小练习—批量读取10X数据
刘小泽写于2020.8.13 主要分为两个部分:前面用linux处理,后面用R处理
下载数据
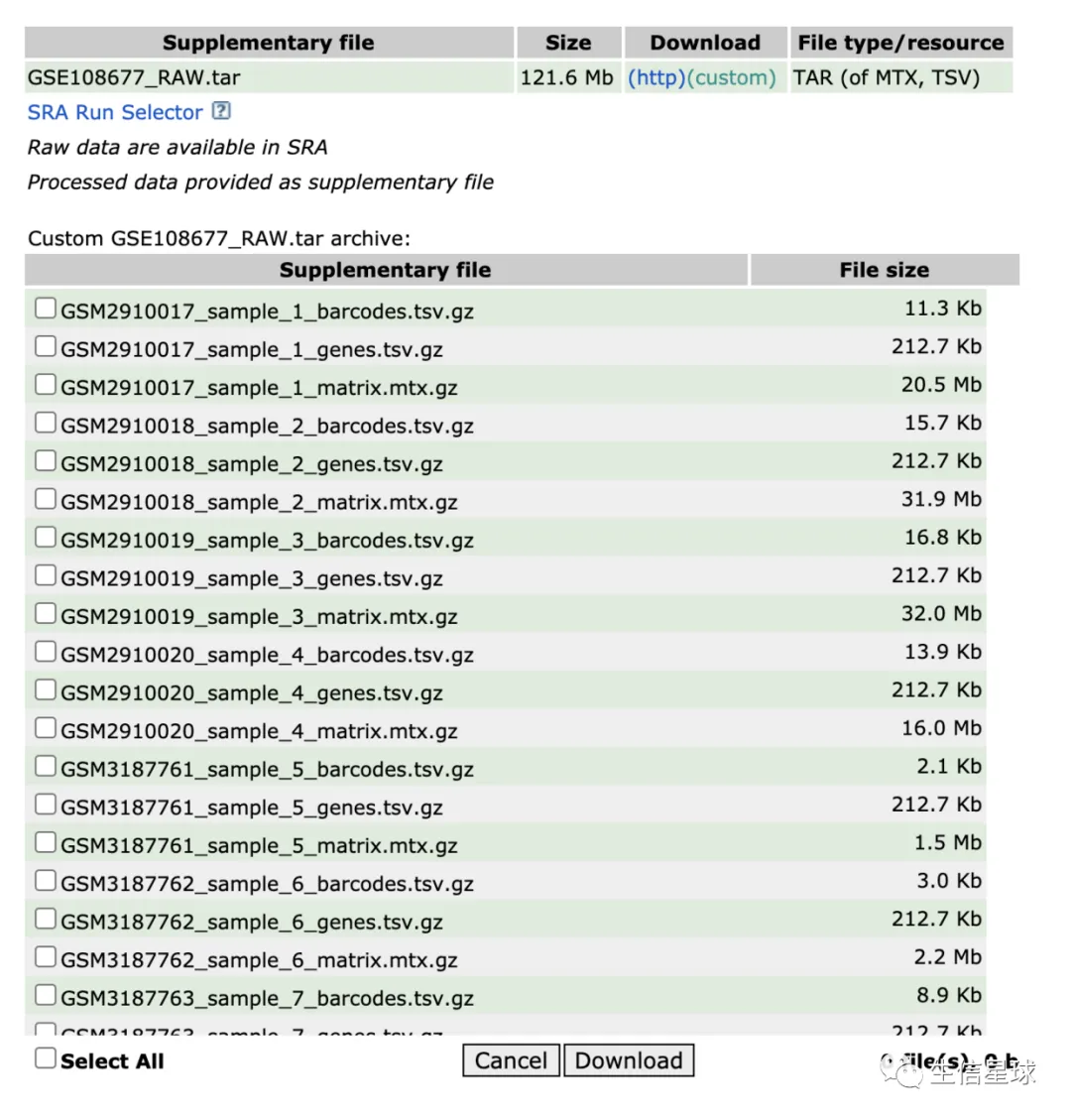
选用的10X测试数据是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108677
数据也可以直接通过网盘下载:https://share.weiyun.com/7fo3qgT5
一共是7个样本:
目的
将这7个样本分别读取,并简单查看一下每个样本的结果如何(比如有多少个细胞,关注的基因出现在哪个细胞等等)
首先是linux部分
**第一步:**先解压,然后最好对每个样本新建一个文件夹
tar -xvf GSE108677_RAW.tar
for i in $(seq 1 7);do mkdir s_${i} ;done
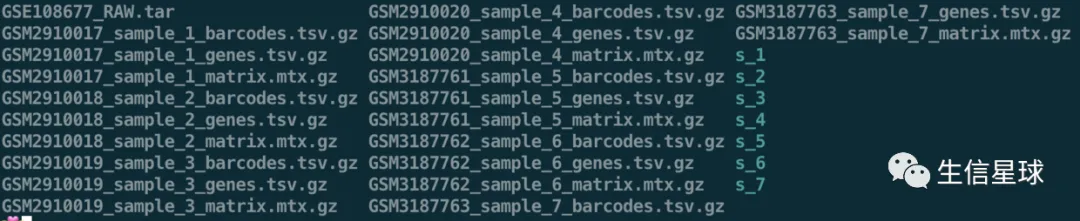
第二步:把各自的10X数据三个必备文件放在对应的文件夹中
for i in $(seq 1 7);do mv *_${i}_*gz s_${i} ;done
# 查看一下
ls s_1
# 每个样本都是这三个文件:barcodes、genes、matrix
GSM2910017_sample_1_barcodes.tsv.gz GSM2910017_sample_1_genes.tsv.gz GSM2910017_sample_1_matrix.mtx.gz
**第三步:**重命名
10X的数据读取是使用Read10X函数,它必须接受特定的命名格式。否则会看到类似下面的报错:
# Error in Read10X(data.dir = dir) :
# Barcode file missing. Expecting barcodes.tsv.gz
它必须要求每个样本都是下面这样的简单命名:

因此,我们需要做的就是:对每个样本文件夹中的每个文件去掉前缀,只保留后面的信息
对于超过三个的数据量,就要用到循环处理
下面的脚本中find是在mac下,如果是linux可能需要稍作调整
# 两个循环嵌套,先找文件夹,再重命名
# ##*_是向后取:取最后一个_后面的部分
# 与之相反是:%%_* 它是向前取:取第一个_前面的部分
for i in $(seq 1 7);do find s_${i}/ -name "*gz" | while read n ;do mv $n s_${i}/${n##*_};done ;done
**第四步:**解压
for i in $(seq 1 7);do gunzip s_${i}/* ;done
接下来是R语言部分
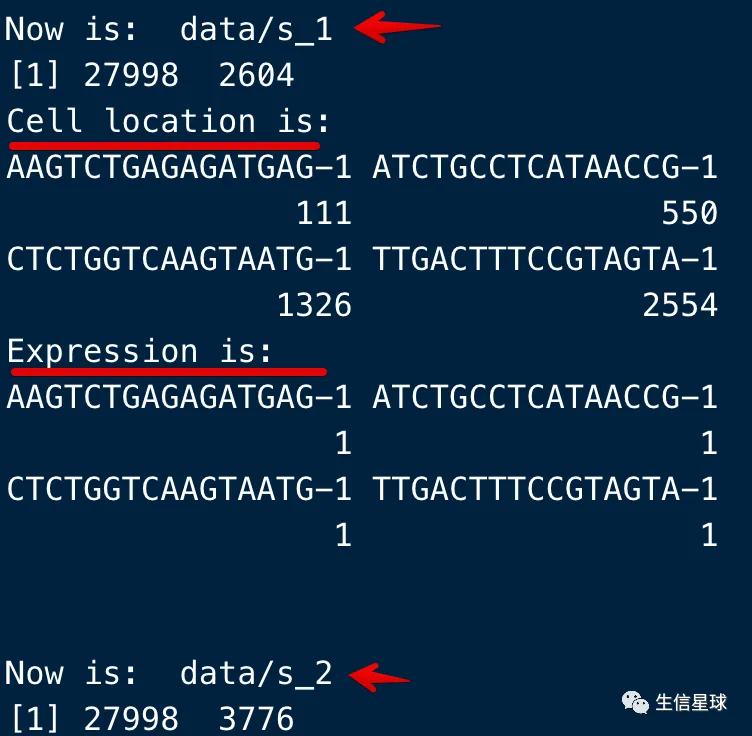
可以简单看看各个样本的细胞数量,以及关注的基因有没有在样本中出现
rm(list = ls())
options(stringsAsFactors = F)
library(Seurat)
# 依然是一个循环
for (i in 1:7){
# i=1
dir=paste0('data/s_',i)
test=Read10X(data.dir = dir)
if('Pigr'%in%rownames(test)){
cat('Now is: ', dir,'\n')
print(dim(test))
cat('Cell location is: ','\n')
print(which(test['Pigr',]!=0)) #Pigr出现在第几个细胞
cat('Expression is: ','\n')
print(test['Pigr',test['Pigr',]!=0]) # Pigr表达量
cat('\n\n')
}
}