202-会了GEO数据下载,来看看怎么上传吧
刘小泽写于2020.8.12
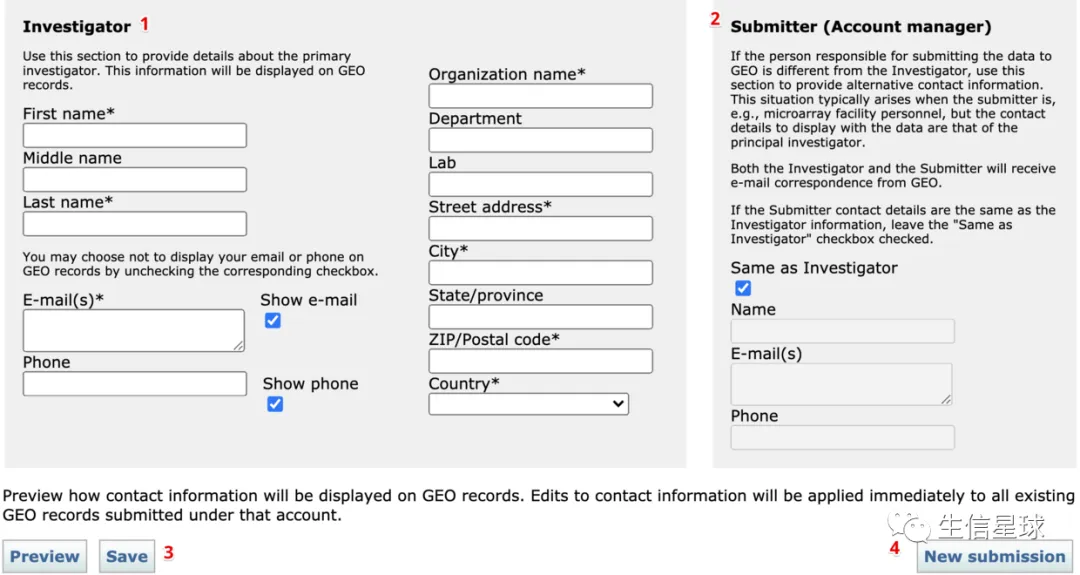
1 注册NCBI GEO账号
先注册NCBI账号,在:https://www.ncbi.nlm.nih.gov/
然后注册GEO账号,在:https://www.ncbi.nlm.nih.gov/geo/submitter/
GEO可上传的数据类型种类主要集中在芯片和高通量数据,比如芯片数据的四大主流:Affymetrix、Agilent、Nimblegen、Illumina,高通量的RNA-Seq、ChIP-Seq、ATAC-Seq等。另外还有RT-PCR、SAGE数据可以上传
2 提交高通量测序数据须知
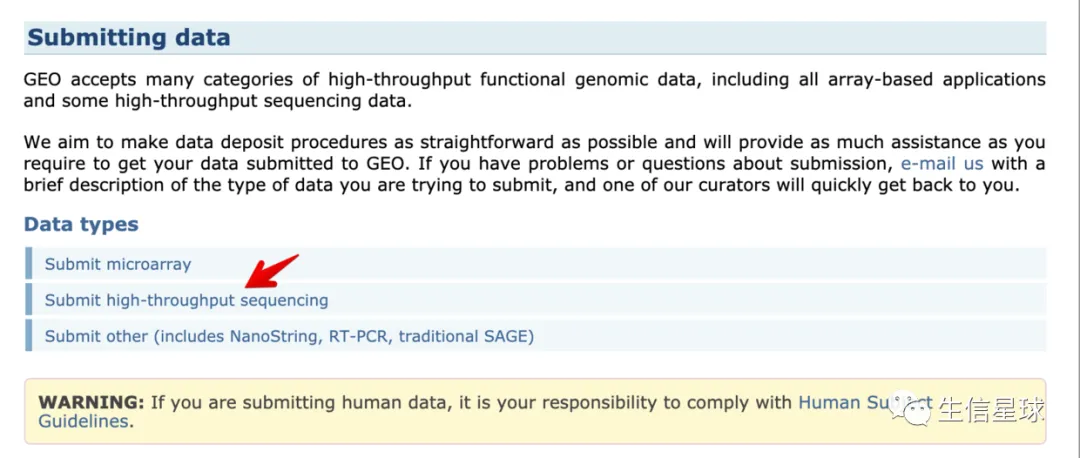
重点需要提交三部分:
- 实验总览(metadata spreadsheet):参考样本 https://www.ncbi.nlm.nih.gov/geo/info/examples/seq_template.xlsx
- 处理过后的数据(processed data files):需要注意
- 不可以提交中间过程的比对文件(如BAM、SAM、BED),但如果结果只有这样的比对文件,可以写信给他们询问是否合格
- 一般来说,提交什么类型的数据取决于实验类型:
- 表达量相关的数据:例如 genes, transcripts, exons, miRNA等表达量,需要原始表达矩阵 或 标准化后的表达矩阵(例如 Cufflinks, Cuffdiff, DESeq, edgeR的结果文件)。可以每个样本一个文件,也可以使用一整个表达矩阵,但需要包含全部基因和全部样本的信息(不可以只用差异基因)
- ChIP-Seq数据:必须包含有关于peak丰度的文件(如WIG, bigWig, bedGraph)
- 所有处理过的文件描述都必须体现在metadata文件中
- 如果提交了WIG, bedGraph, GFF, GTF文件,格式需要参考: UCSC file format FAQ
- 原始数据(raw data files): GEO的原始数据也是会提交给SRA
- 必须是包含reads、质量值的原始fastq格式,不符合要求的数据会直接从GEO系统中删除
- 如果测序数据使用了barcode(例如10x Genomics, Drop-Seq, InDrops的数据),可以提交不经过拆分的multiplexed files;对于其他多路复用(Multiplexed)的数据来说,必须要先经过demultiplex操作,将样本分开
- PE测序数据:一般每个run会产生两个数据(特殊情况下,每个run中的序列和质量值文件是分开的,也就是产生了4个文件)
- MD5Sum:推荐使用MD5验证数据,方法是:
- Unix:
md5sum <file> - OS X:
md5 <file> - Windows: 需要用某些应用程序(如
winmd5free,或者 Microsoft’s File Checksum Integrity Verifier (FCIV) utility ))
- Unix:
- 关于数据压缩:为了加快传输,可以适当将数据压缩,但不强求。可以使用gzip、bzip2(后缀是
.gz或.bz2) ,但不要压缩二进制文件(如BAM、bigWig、bigBed),也不要上传ZIP文件
3 GEO接受的数据与不可接受的数据
GEO可接受的
基因表达、基因调控、表观以及其他功能基因组学研究,例如
- mRNA profiling, RNA-seq ( example)
- small RNA profiling, miRNA-seq ( example)
- ChIP-Seq ( example)
- HiC-seq ( example)
- methyl-seq, bisulfite-seq ( example)
GEO不可接受的
需要权限访问的人类数据:可以提交给 dbGaP and controlled access SRA
转录本组装:可以提交给 SRA 以及 Transcriptome Shotgun Assembly Database)
宏基因组测序:可以提交给 SRA
重测序以及变异相关研究:可以提交给 SRA 或 合适的 variation resource
全外显子数据:可以提交给 SRA
4 重头戏-实验总览(metadata spreadsheet)
参考样本: https://www.ncbi.nlm.nih.gov/geo/info/examples/seq_template.xlsx
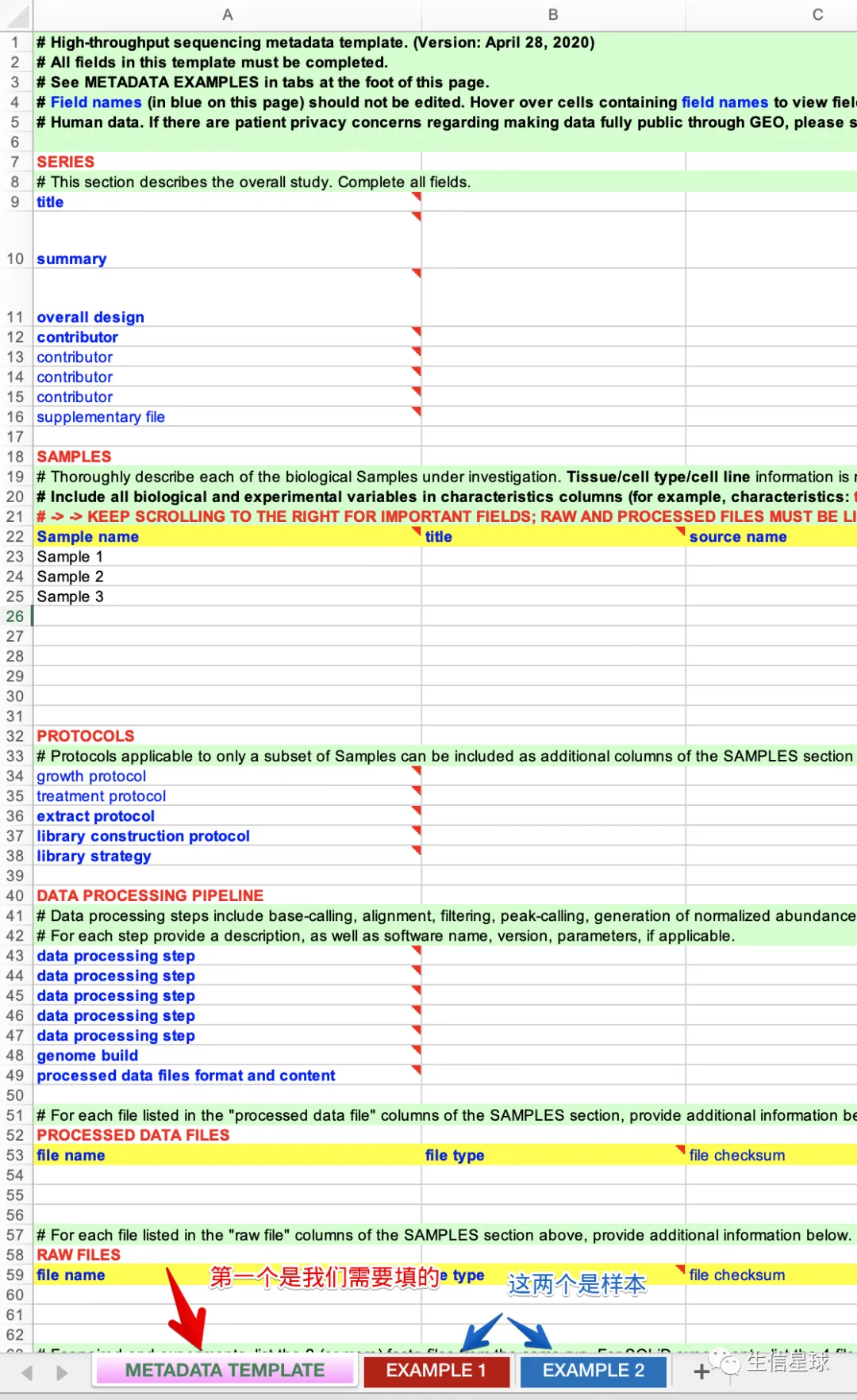
总共包含7大块
4.1 SERIES
与文章相关的内容
- 标题(title)
- 摘要(summary)
- 实验设计(overall design)
- 参与者(contributor):可以写多个
- 附件(supplementary file)

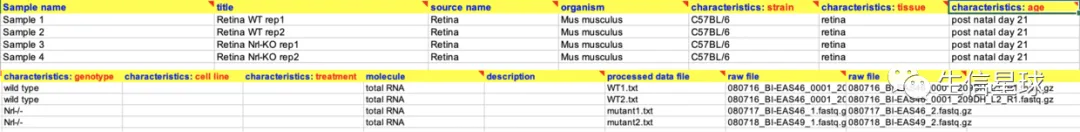
4.2 SAMPLES
与样本信息相关的内容
- 样本编号(Sample name)
- 样本名称(title)
- 样本来源(source name)
- 物种(organism)
- 样本描述(characteristics: strain、tissue、age、genotype、cell line、treatment)
- 与该样本相关的文件(molecule、processed data file 、raw file)
4.3 PROTOCOLS
样本的实验操作以及建库流程,简单描述即可

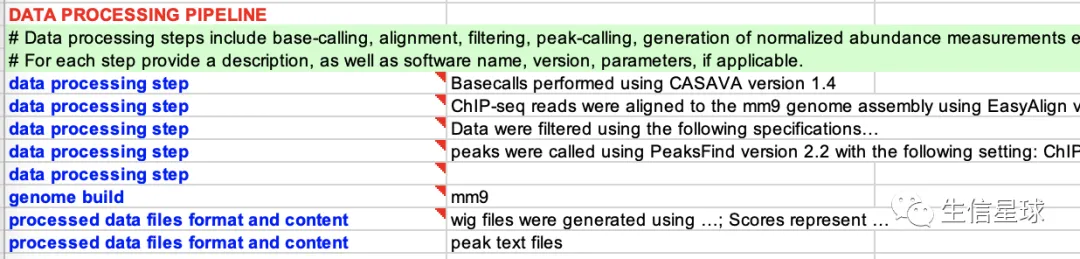
4.4 DATA PROCESSING PIPELINE
数据处理描述,比如基因组版本是什么、怎么比对、怎么过滤、怎么找peaks、怎么定量
4.5 PROCESSED DATA FILES
数据处理后的文件名称
- file name
- file type:除了raw count数据,其余可以统一写成abundance measurements
- file checksum

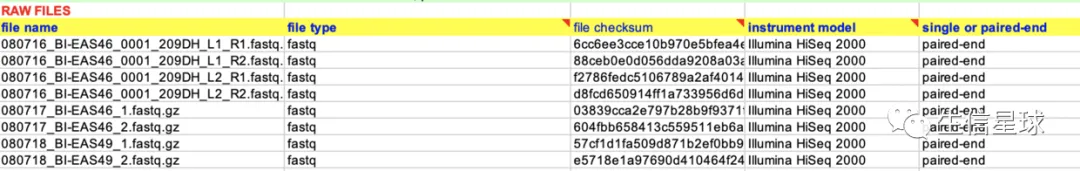
4.6 RAW FILES
- 原始数据名称(file name)
- 文件类型(file type):比如fastq
- md5校验(file checksum)
- 测序仪器型号(instrument model)
- 单端or双端(single or paired-end)
4.7 PAIRED-END EXPERIMENTS
如果使用了双端测序数据,需要列出各自的名称
- file name 1
- file name 2
5 准备工作结束后,可以开始上传
上面的实验总览(metadata spreadsheet)、处理过后的数据(processed data files)、**原始数据(raw data files) ** 都准备好,就可以开始准备上传了
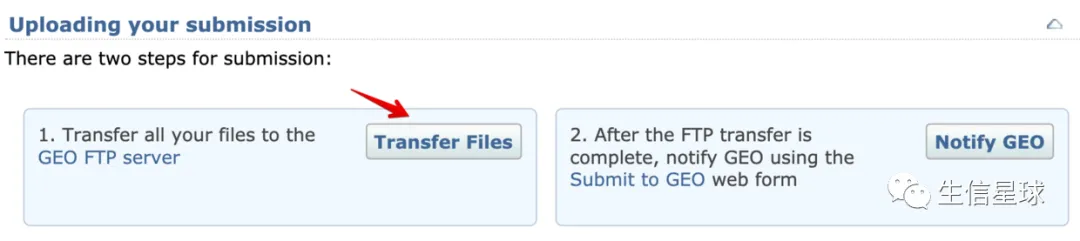
学习如何使用FileZilla进行上传
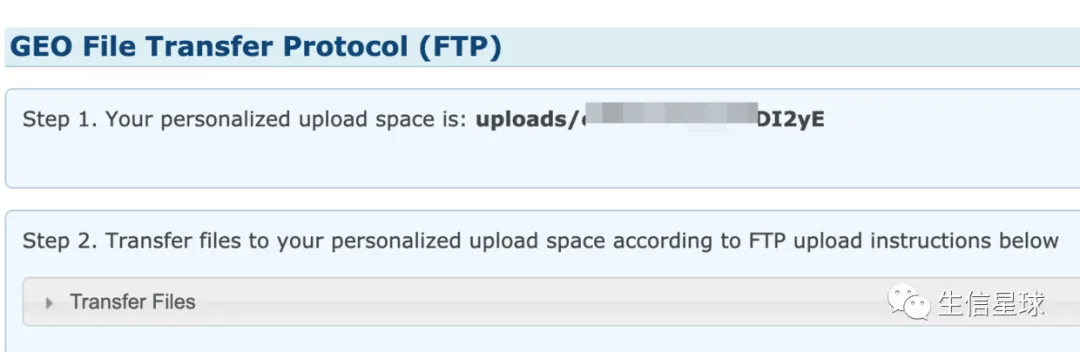
首先会看到自己的上传目录,一会将用到
然后设置FileZilla:
- host (ftp-private.ncbi.nlm.nih.gov)
- username (geoftp)
- password (rebUzyi1)
此时会发生报错,忽略它
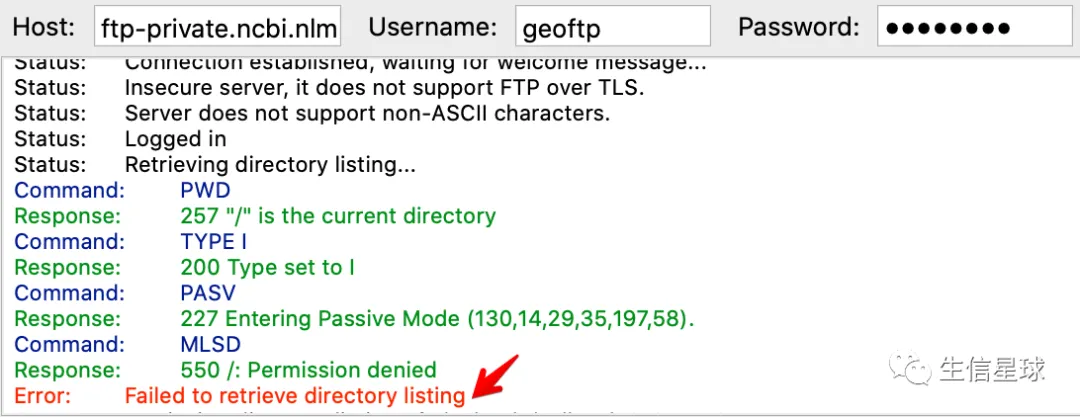
修改Remote site,然后回车连接:
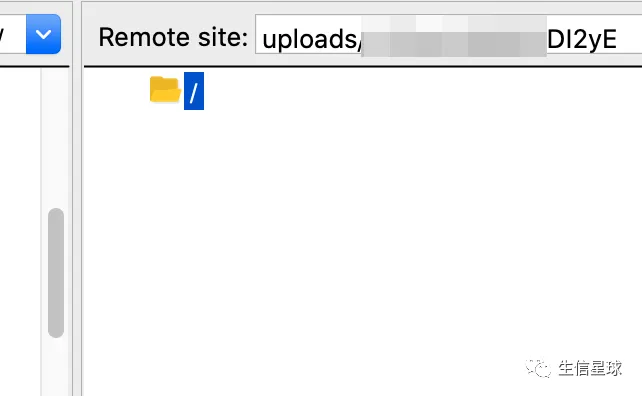
最后就可以将本地数据上传到GEO指定位置了
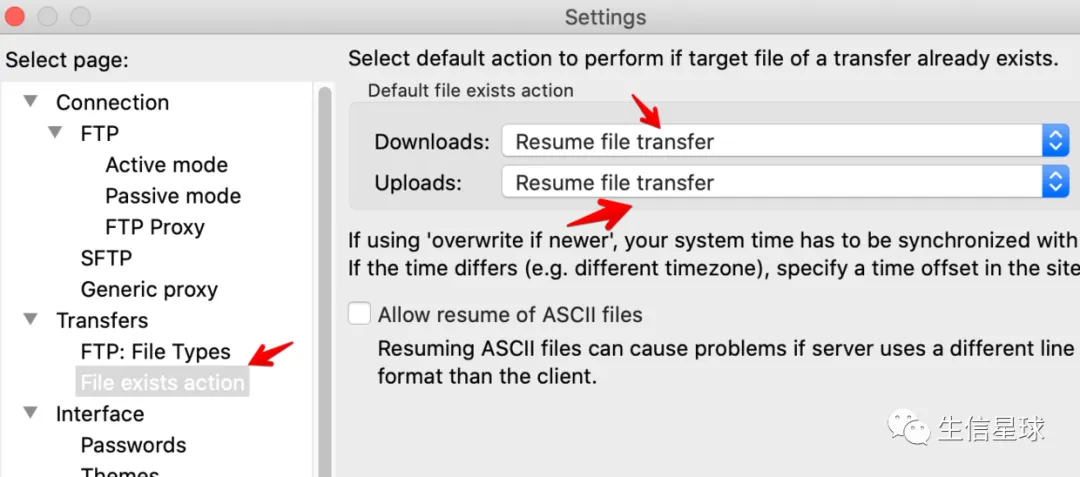
Tips:为了避免FileZilla上传过程出现中断,可以设置断点续传
6 最后,提醒GEO数据上传完成
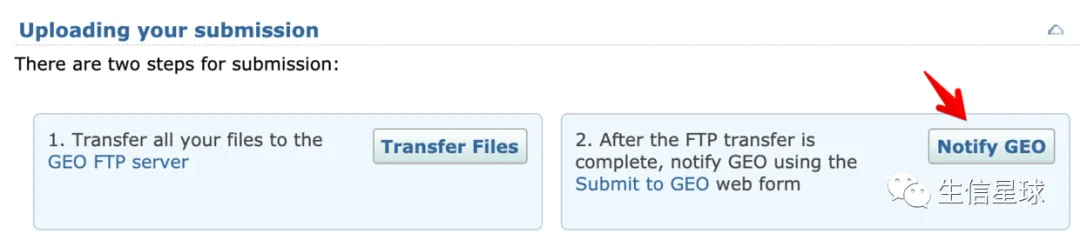
并且会提示再核实一遍信息,没有问题的话5个工作日内就会进行审核
Tips:补充
来自:https://www.ncbi.nlm.nih.gov/geo/info/submissionftp.html
在上传前,首先新建一个主目录,例如:
geo_submission_august17,然后把所有的要传送的数据都放里面。如果paper中包含了多个组学数据(例如同时做了ChIP-Seq、RNA-Seq),可以在主目录下新建不同组学的子目录,例如:geo_submission_august17/ChIPseq如果总文件大小超过了1T,需要提前联系GEO
对于Mac、PC用户,推荐使用客户端,如Filezilla;对于LINUX/UNIX用户,推荐使用
ncftp、lftp命令GEO的FTP server登录:

登录后,一定要去到自己的上传目录下,再将主目录拖拽上传
最后通知GEO的时候,也要提醒他们自己上传的目录名字是什么【在数据传完之前,不要通知GEO】。如果不通知,两个星期后数据就会被删除
文件可以压缩为gz或bzip2【但二进制文件不能压缩】,也可以用tar打包,但千万不要用ZIP
文件名中只能存在:
alphanumerals [A-Z, a-z, 0-9], underscores [_] and dots [.]