200-求助效应成功案例之--如何去掉匹配字符后的一连串
刘小泽写于2020.7.13 名词解释:“求助效应”——来自花花。 意为:当自己有一个问题想要问别人,但还没得到他人答复时,自己就会在等待期间自行探索,最终先于他人,自行解答成功
前言
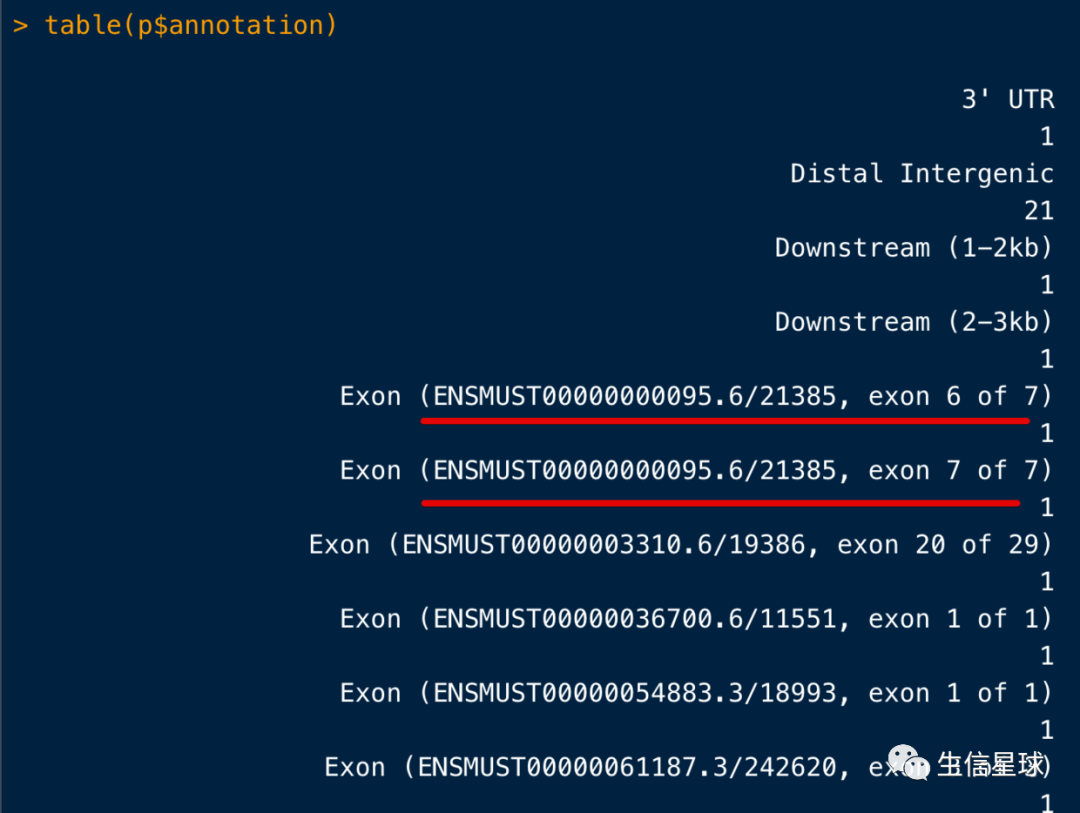
今天早上想要统计一下ChIPseeker的peaks注释结果,但是table一下看到下面的情况,本来就只是想看看有多少个exon,不想看这么详细。所以我想把Exon之后的字符串(ENSMU* 去掉再重新统计【当然这个问题也存在与intron中】
简单尝试了一下,发现不能成功匹配正则表达式,于是想着问问花花吧
五分钟后,花花没回复。。。
当然,期间我自己进行了不情愿的尝试 本来以为是一个小问题而已,就不想动脑了。但事实证明还得靠自己搜索试错解决
尝试第一次
我搜索的条件是:r remove character from a pattern
结果就立刻找到了答案:https://stackoverflow.com/questions/11776287/remove-pattern-from-string-with-gsub

以为加上这个参数就会匹配到
> head(table(gsub("\\(ENSMUST*", "", p$annotation,perl = T)))
3' UTR Distal Intergenic
2 34
Exon 00000003310.6/19386, exon 20 of 29) Exon 00000005066.8/26395, exon 2 of 11)
1 1
Exon 00000005066.8/26395, exon 3 of 11) Exon 00000005066.8/26395, exon 7 of 11)
1 1
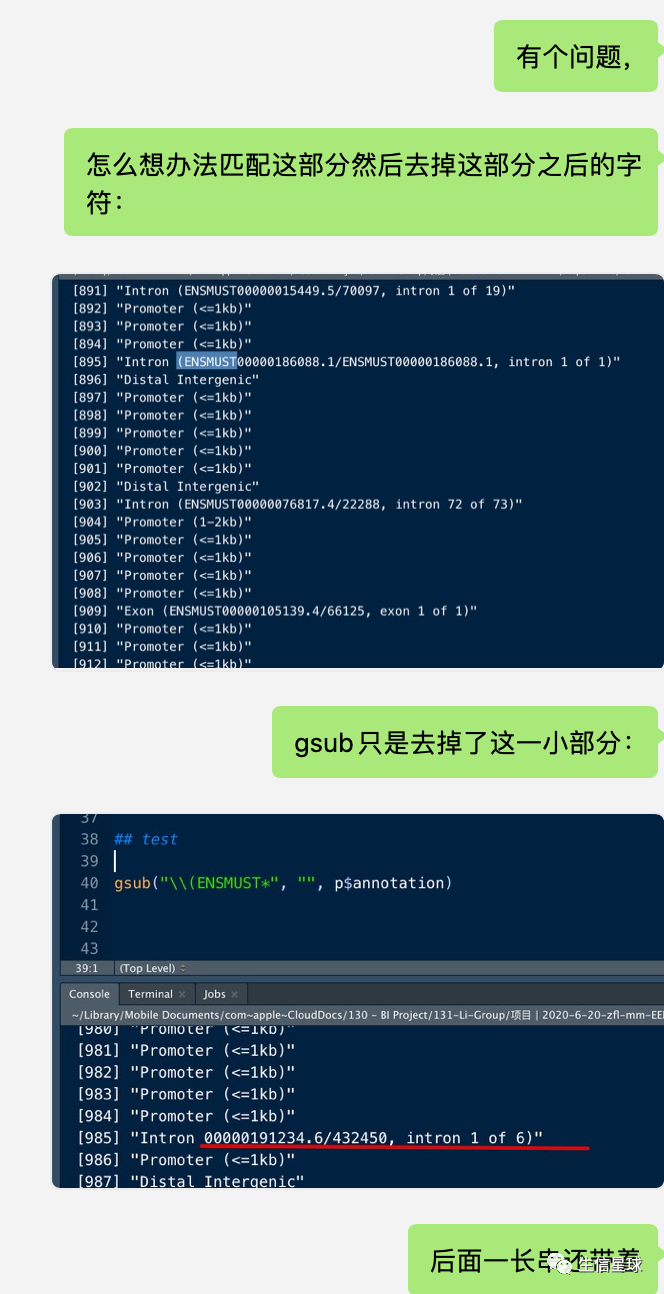
但事实是,这个依然只是去掉了(ENSMUST这一小部分,后面的没去掉。
因此,这个参数加不加都一样
尝试第二次
我重新思考了一下正则表达式真的写对了吗?
我的需求是:匹配(ENSMUST后面的一串字符,但是我之前写的是:(ENSMUST*
在正则表达式中,有这么几条常用的规定:

因此,看到这里大家想必都明白了,我的表达式没写对
(ENSMUST* 只是表示:匹配前一个T字母出现多次或0次,它匹配到的其实是:ENSMUSTTTTTTT这样的
最后确定,我少了一个. 号。看来真的是对正则表达式生疏了

好至此匹配的问题解决
再回到ChIPseeker这里
其实之前在写: 一起学习一遍ChIPseeker的使用 这一篇的时候就提到了:
可以在peak注释之前就设置好,省的下面再删减字符串
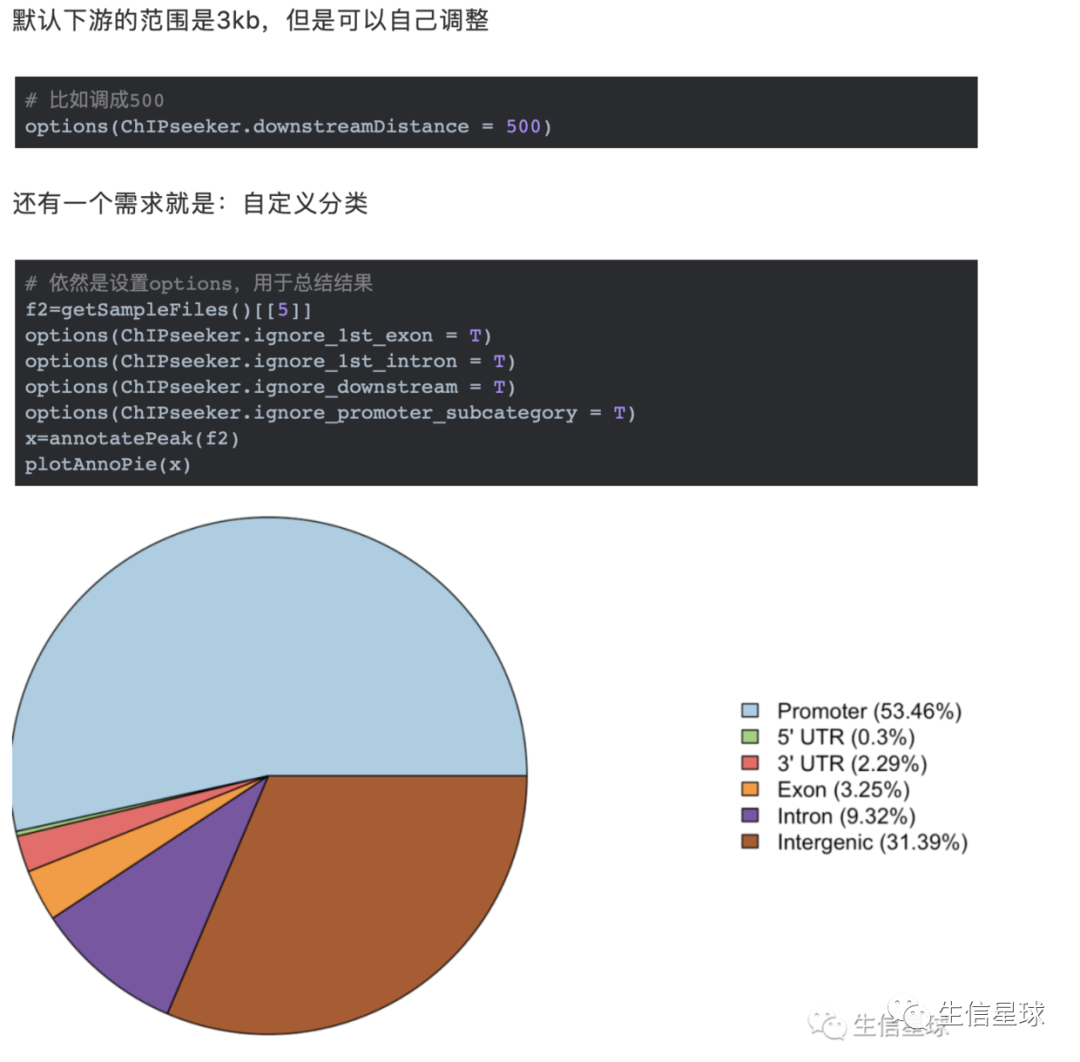
注释结果中的分类有下游(Downstream,默认范围3kb)但没有上游,这是因为Promoter定义为了转录起始位点(TSS)的上下游区域,包含了上游;另外这个下游是是基因间区的一部分,更确切是指紧接着基因的下游;这里的上游和下游其实都是基因间区,独立出来是因为和基因直接连接,是很近的区域=》近端基因间区
当然,基因间区还包含更远的间区(Distal intergenic)=》远端基因间区
最后,如何实现批量操作
很多生信操作的最终目的都是方便批量处理,这里的peaks注释也不例外
比如我们可以简单看一个peak注释文件的类型统计,但是如果有几十个呢?
循环走起!
files=list.files(pattern = '*peakAnno*',path = '.', recursive = T,full.names = T)
for (filename in files){
# filename=files[1]
prefix=sapply(strsplit(filename, '_'), function(x) paste(x[2:4], collapse = '-'))
cat(paste0('Peak types for ',prefix,'\n'))
p=read.csv(filename,row.names = 1)
cat(names(table(gsub("\\(ENSMUST.*", "", p$annotation))),'\n')
cat(table(gsub("\\(ENSMUST.*", "", p$annotation)),'\n\n')
}

这样可以看一眼,但还是感觉不如数据框更直观,而且我们想给别人看,也是要发给他们表格。
下一个问题就是:如何变成数据框?
目的就是得到下图:
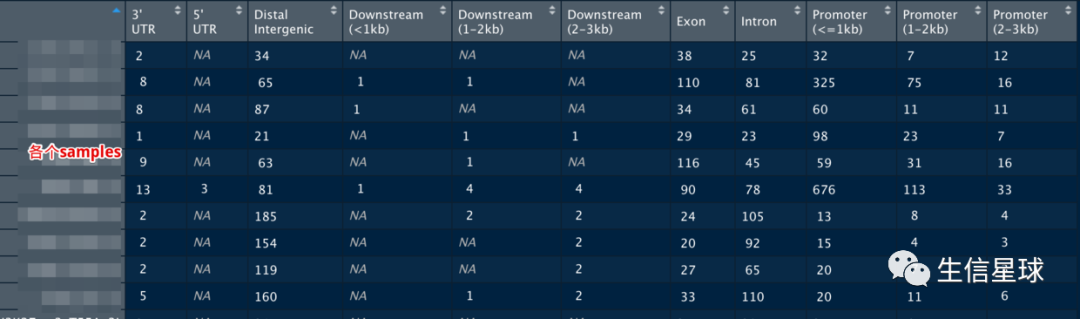
这里需要注意,不是每一个peak注释结果的列数都是固定的,也就是说,有的结果可能只有3‘UTR peaks,有的可能5’和 3‘都有。这就给数据框组合带来了困扰
这里最好先按列合并,比如merge,left_join都对列的合并有比较好的支持,而行合并可能问题多多
rm(list = ls())
options(stringsAsFactors = F)
require(stringr)
library(tidyverse)
# 新建一个空数据框
dat=data.frame(c("3' UTR","5' UTR","Distal Intergenic", "Downstream (<1kb)",
"Downstream (1-2kb)", "Downstream (2-3kb)", "Exon",
"Intron", "Promoter (<=1kb)", "Promoter (1-2kb)", "Promoter (2-3kb)"))
colnames(dat)='type'
files=list.files(pattern = '*peakAnno*',path = '.', recursive = T,full.names = T)
# 一个循环,有匹配的就把数字填进去,没有匹配就NA(left_join的功能)
for (n in 1:length(files)){
# n=1
# n=2
prefix=sapply(strsplit(files[n], '_'), function(x) paste(x[2:4], collapse = '-'))
# cat(paste0('Peak types for ',prefix,'\n'))
p=read.csv(files[n],row.names = 1)
# cat(names(table(gsub("\\(ENSMUST.*", "", p$annotation))),'\n')
# cat(table(gsub("\\(ENSMUST.*", "", p$annotation)),'\n\n')
pd=as.data.frame(table(gsub(" \\(ENSMUST.*", "", p$annotation)))
colnames(pd)=c('type',prefix)
pd$type=as.character(pd$type)
dat=left_join(dat,pd,by='type')
}
# 最后转置回来
final=as.data.frame(t(dat))
colnames(final)=final[1,]
final=final[-1,]