024-表达矩阵的合并
刘小泽写于2018.8.19
实例测试(单行perl结合R语言)
现在有三个count文件,SRR3589956.count、SRR3589957.count、SRR3589958.count
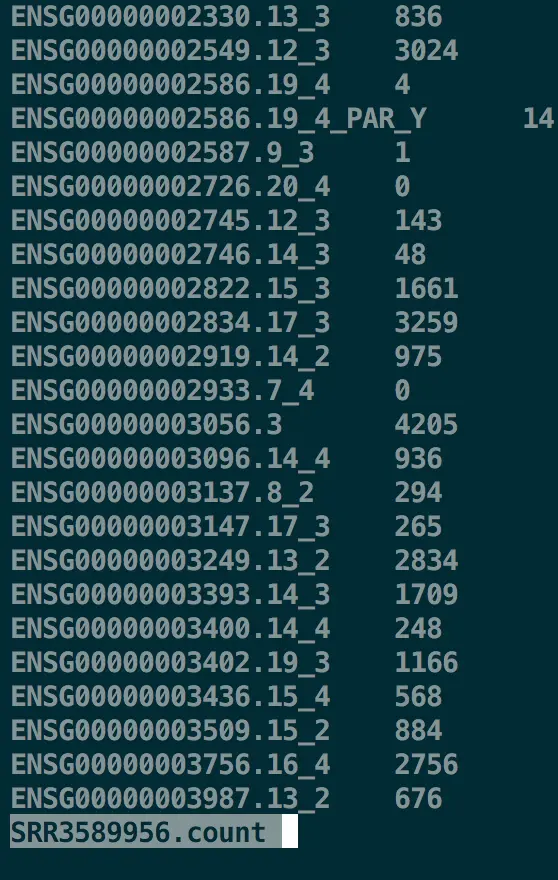
第一步 初步合并各个计数文件
先将两个文件合并,第一列是样本名,第二列是基因名,第三列是count结果
perl -lne 'if ($ARGV=~/(.*).count/){print "$1\t$_"}' *.count >matrix.count
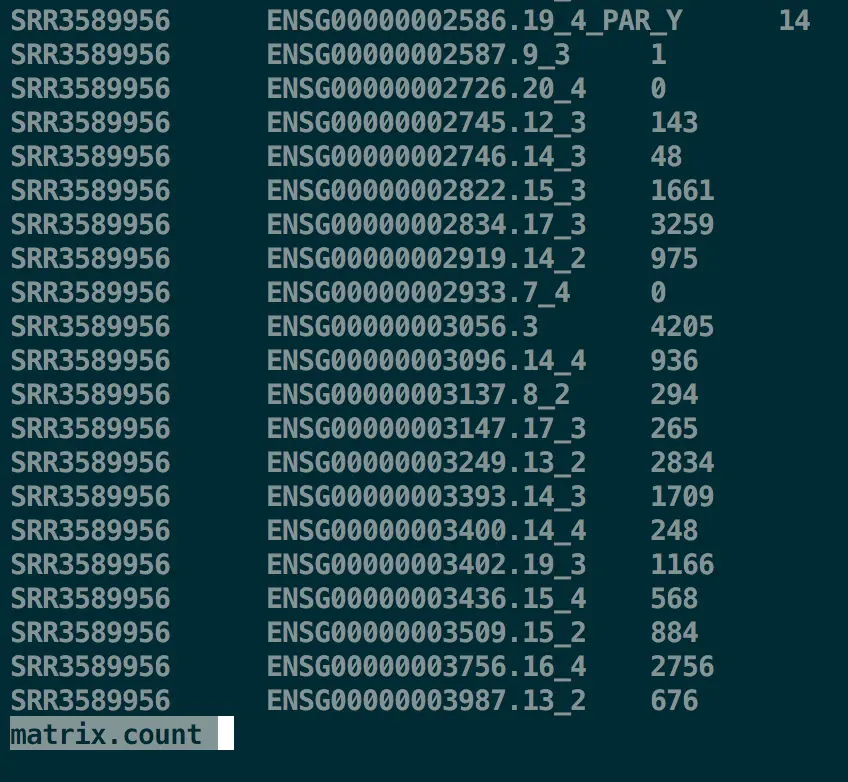
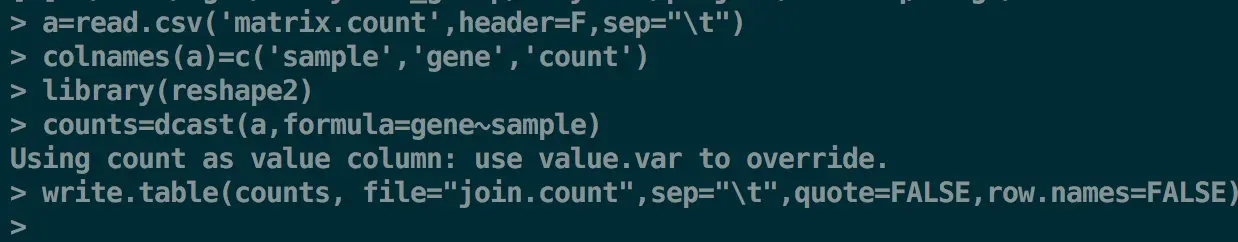
第二步 R重塑合并的计数矩阵
a=read.csv('matrix.count',header=F,sep="\t")
colnames(a)=c('sample','gene','count')
library(reshape2)
counts=dcast(a,formula=gene~sample)
write.table(counts, file="join.count",sep="\t",quote=FALSE,row.names=FALSE)
可以直接在命令行运行(前提是安装了R)
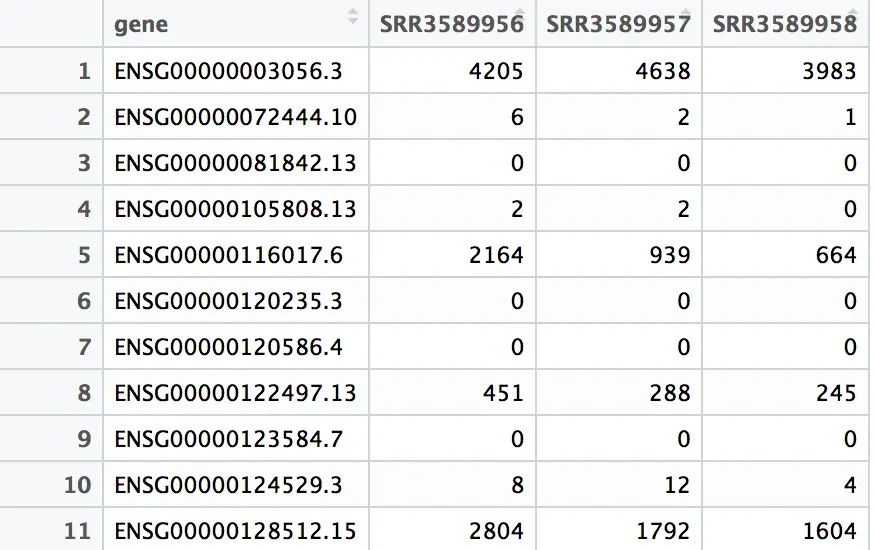
第三步 探索
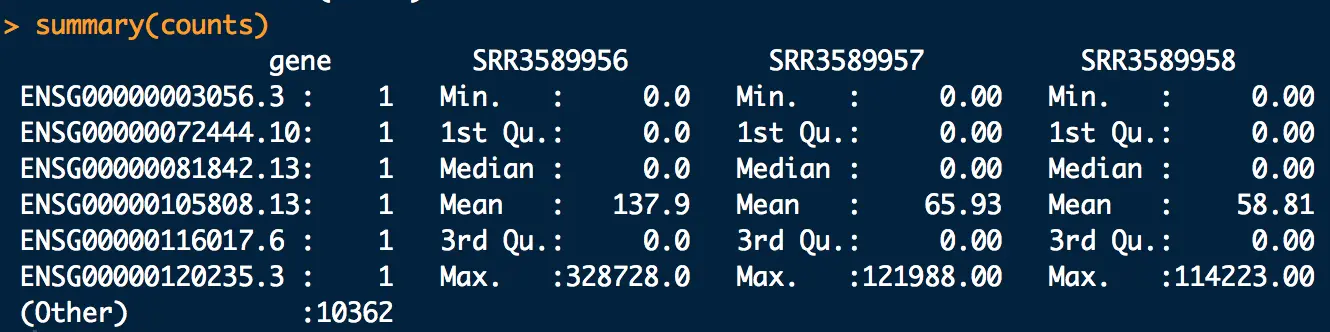
- 统计各个样本count值(最值、中位数)
按照基因来统计counts的平均数
install.packages("dplyr") library(dplyr) gene_mean = group_by(a,gene)%>%summarize(mean=mean(count)) #另外也可以按照sample来统计 #当然,除了mean平均数,还可以求中值median,最值max、min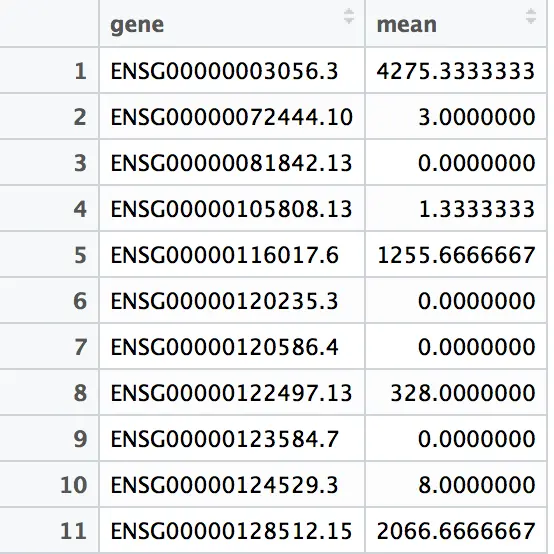
(没有实际数据)自己生成测试数据
目的:生成a-g.txt 7个文件,每个文件中第一列为基因名,从gene_1到gene_99,第二列是表达量,1000以内随机整数
#新建测试文件test.sh
perl -le '{print "gene_$_\t".int(rand(1000)) foreach 1..99}'
# -e后面紧跟着引号里面的字符串是要执行的命令;
# -l输出换行
#想要输出gene_1这样类型的,gene_后面的数字用一个变量$_代替,这个变量相当于占一个位置,它的赋值是在后面foreach 1..99,表示 $_从1到99
#加一个\t表示一个tab键,空4格
#rand生成随机数,int限制整数
#注意到"gene_$_\t"与后面生成整数函数之间有一个.【不加.就会报错,它的作用应该是分离字符串和函数】
#perl脚本调用test.sh
perl -e 'system("bash test.sh > $_.txt") foreach a..g'
#结果就生成了想要的测试文件
开始统计:
#先合并
perl -lne 'if ($ARGV=~/(.*).txt/){print $1\t$_}' *.txt > matrix.count
#进入R studio
#然后重复上面👆第二步