189-Bowtie2比对过程中发生的一个小错误
刘小泽写于2020.5.21 记录一下踩的坑
上报错
手上有一些ATAC-seq的双端测序数据(25bp)进行了fastp质控过滤后准备进行比对,结果比对过程发生报错,信息如下
# Bowtie2
Error, fewer reads in file specified with -1 than in file specified with -2
terminate called after throwing an instance of 'int'
(ERR): bowtie2-align died with signal 6 (ABRT) (core dumped)
貌似显示双端测序数据中两个文件reads数不等
如何查看测序数据的信息
简单使用脚本
zcat raw/test_1.fastq.gz | wc -l| awk '{print $1/4}'
# 4237351
zcat raw/2d_2.fastq.gz | wc -l| awk '{print $1/4}'
# 4237351
使用小工具
seqkit stat raw/test_1.fastq.gz raw/test_2.fastq.gz
# file format type num_seqs sum_len min_len avg_len max_len
# raw/test_1.fastq.gz FASTQ DNA 4,237,351 105,933,775 25 25 25
# raw/test_2.fastq.gz FASTQ DNA 4,237,351 105,933,775 25 25 25
看到原始数据的两个文件中reads数相等
既然原始数据一致,那么质控过滤后的呢?
果然,利用seqkit stat发现clean的一对PE数据中reads数不一致
看了一下如何进行过滤的
sample=test
fq1=~/atac/raw/${sample}_1.fastq.gz
fastp -i $fq1 -o clean/${sample}.fq1.gz \
-I $fq2 -O clean/${sample}.fq2.gz \
--thread=4 --length_required=10 --n_base_limit=5 \
--compression=6 -R ${sample}_1 1>log/log.fastp.${sample}.1.txt 2>&1
fq2=~/atac/raw/${sample}_2.fastq.gz
fastp -i $fq2 -o clean/${sample}.fq2.gz \
--thread=4 --length_required=10 --n_base_limit=5 \
--compression=6 -R ${sample}_2 1>log/log.fastp.${sample}.2.txt 2>&1
忽然想起来,这个脚本是从ChIP-seq脚本中复制粘贴过来的,只改了对应的fq1和fq2,但一般ChIP-seq都是单端的,所以这个fastp就是对单端数据的过滤
问题的根源在于:这里把双端数据当成了两个单端分别进行处理,那么结果就不一致啦
将代码修改为双端对应的就好了
# 如果是双端,就需要指定-i -o (for input 1)
# -I -O (for input 2)
sample=test
fq1=~/atac/raw/${sample}_1.fastq.gz
fq2=~/atac/raw/${sample}_2.fastq.gz
fastp -i $fq1 -o clean/${sample}.fq1.gz \
-I $fq2 -O clean/${sample}.fq2.gz \
--thread=4 --length_required=10 --n_base_limit=5 \
--compression=6 -R ${sample} 1>log/log.fastp.${sample}.txt 2>&1
收获
一般来说,软件的报错信息都会显示在前面,需要观察加发散思维
例如
# bowtie2
Error: reads file does not look like a FASTQ file
terminate called after throwing an instance of 'int'
(ERR): bowtie2-align died with signal 6 (ABRT) (core dumped)
看到这里,就要去看原始的fq文件(主要信息就是第2行和第4行),可能存在长度不一致的情况
zcat fastq.gz | paste - - - - | awk -F"\t" '{ if (length($2) != length($4)) print $0 }'
# 如果要挑出来这些reads
zcat fastq.gz | paste - - - - | awk -F"\t" '{ if (length($2) != length($4)) print $0 }' | tr '\t' '\n' > error_reads.fastq
再例如,一个R语言的报错
安装rMATS时看着满屏的报错,眼花缭乱,但后面其实都在重复说找不到libgsl.so.0这个库

于是关键词搜索一下libgsl.so.0 rmats:https://www.biostars.org/p/336864/
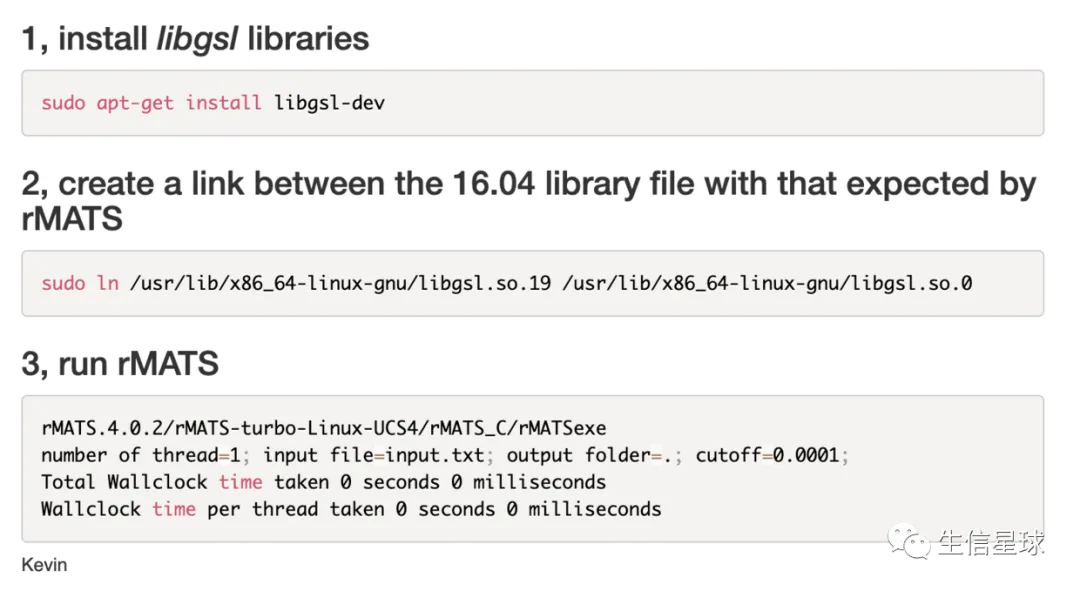
或者使用:sudo apt-get install libgsl0-dev (https://cloud.tencent.com/developer/article/1366294)