188-继续来看pheatmap那些有趣的事情
刘小泽写于2020.5.18 上次看了 热图如何去掉聚类树的同时保留聚类的顺序 ,有朋友留言给出了其他的解决方法,非常感谢!文中会给出答案 另外,这次还是带着数据,去看看pheatmap其他的有用操作
上数据!
test = matrix(rnorm(200), 20, 10)
test[1:10, seq(1, 10, 2)] = test[1:10, seq(1, 10, 2)] + 3
test[11:20, seq(2, 10, 2)] = test[11:20, seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20, seq(2, 10, 2)] + 4
# 20行(基因)10列(样本)
colnames(test) = paste("Test", 1:10, sep = "")
rownames(test) = paste("Gene", 1:20, sep = "")
# 和上次不同的是,我们这次设置一个极值(为了后面演示scale)
test[10,10]=100
> rownames(test)[10];colnames(test)[10]
[1] "Gene10"
[1] "Test10"

画图
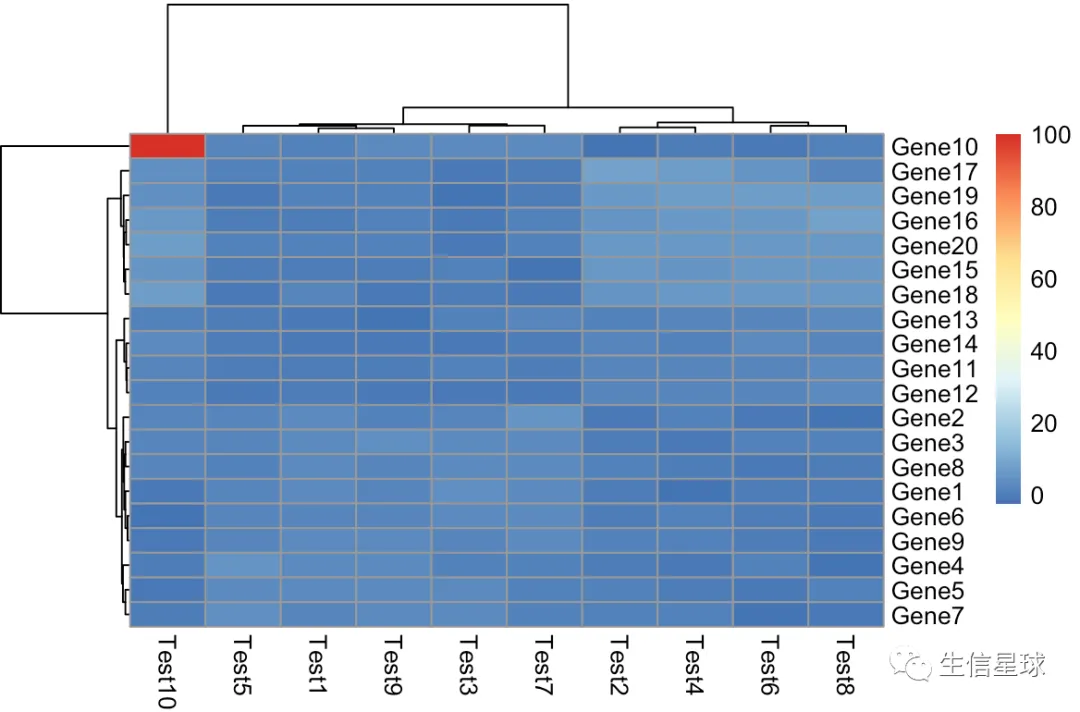
第一个原始图
可以看到Gene10 Test10这里的100大大超过了一般水平,导致画的图受它的影响极大,变得看不出整体的变化趋势
pheatmap(test)
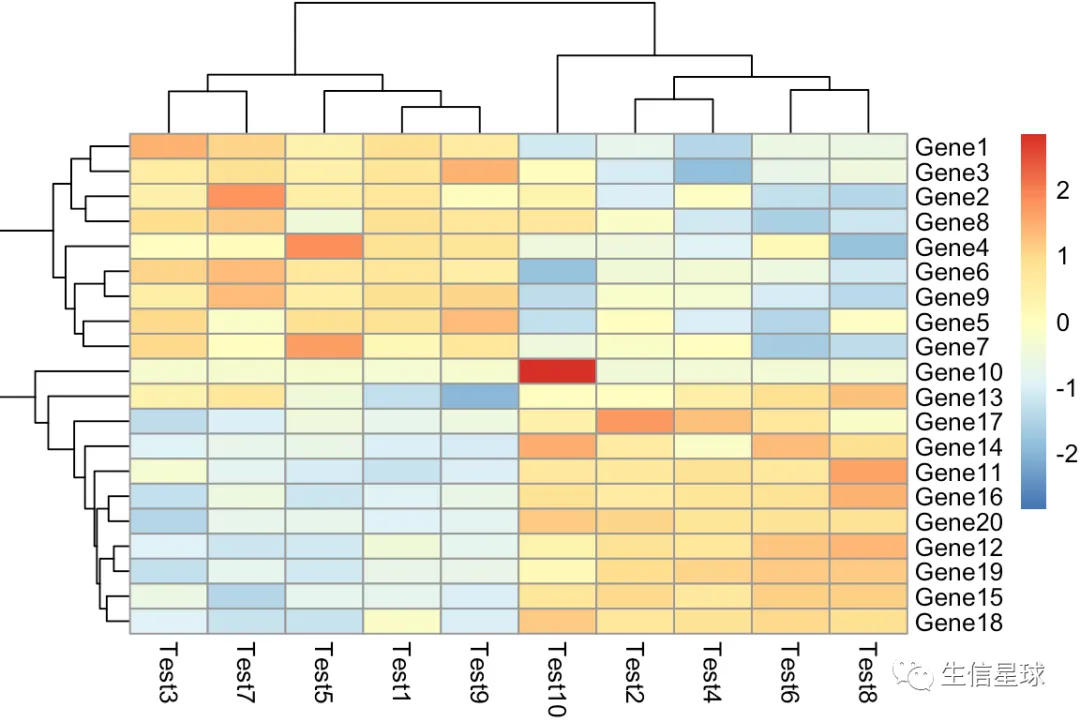
第二个:增加z-score参数的图
设置一个参数scale = 'row' 就会对行进行z-score标准化
什么是z-score? 看个公式就明白了(表达量-均值)/标准差,目的就是不让数据受极值的影响太大。另外z-score一般还会设置一个最大最小值(例如
-2 ~ 2、-1 ~ 1) Z score = (expression G - mean expression G1..Gn)/SD G1..Gn where G is any gene in the pathway and G1..Gn represent the aggregate measure of all of the genes. Cite:Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5(2):73‐81. doi:10.1016/S1525-1578(10)60455-2

pheatmap(test,scale = 'row')
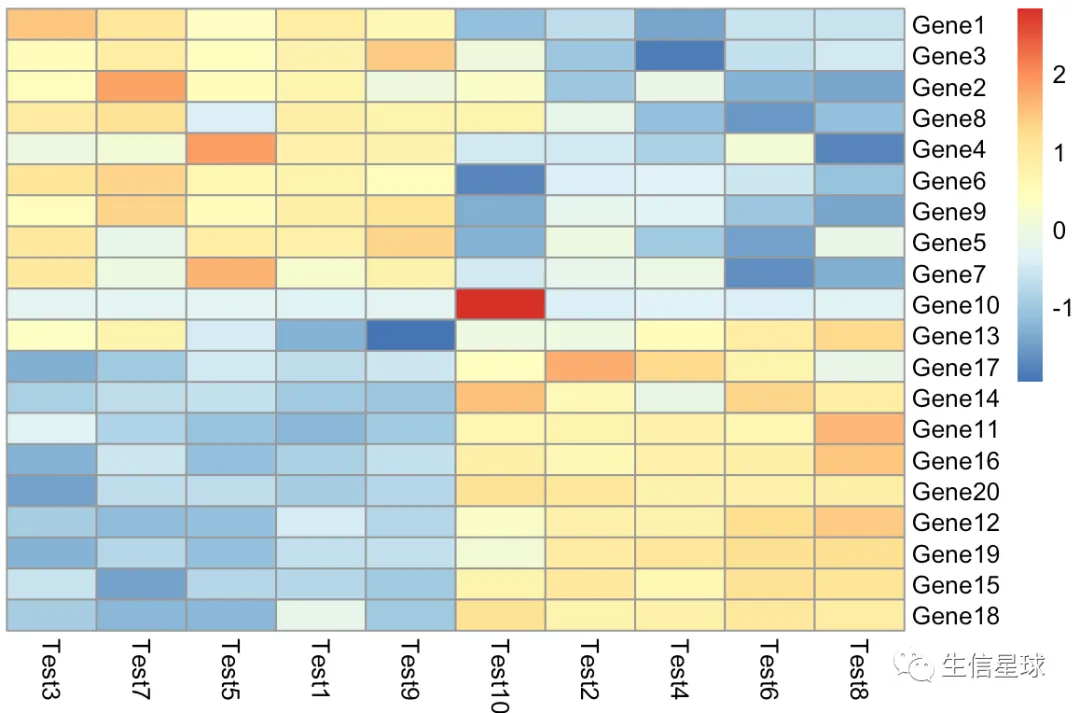
第三个图:手动zscore
如果想要理解某个参数做了什么,就自己模拟这个参数吧
我们这里也来模拟一下这个参数做了什么【并且用热心朋友推荐的方法去掉聚类树】
cal_z_score <- function(x){
(x - mean(x)) / sd(x)
}
diy1 <- t(apply(test, 1, cal_z_score))
# 很巧妙,设置高度为0,就不显示了
pheatmap(diy1,treeheight_row = 0,treeheight_col = 0)
第四个图:聚类是怎么聚的呢?
使用参数的话,很简单:
pheatmap(diy1,clustering_method = 'complete',cluster_rows = T, cluster_cols = T,
clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean")
这个method是直接使用的hlust计算的,全部聚类方法包括:
"ward.D", "ward.D2", "single", "complete", "average" (= UPGMA), "mcquitty" (= WPGMA), "median" (= WPGMC) or "centroid" (= UPGMC).
距离使用dist计算,默认使用欧式距离,全部距离包括:
"euclidean", "maximum", "manhattan", "canberra", "binary", "minkowski"
提取聚类结果:
p1 <- pheatmap(diy1, silent = TRUE) # 只输出结果,不画图
> names(p1)
[1] "tree_row" "tree_col" "kmeans" "gtable"
# 做个丑陋的聚类图看看
library(dendextend)
p1$tree_row %>%
as.dendrogram() %>%
plot(horiz = TRUE)
# 聚类基因名
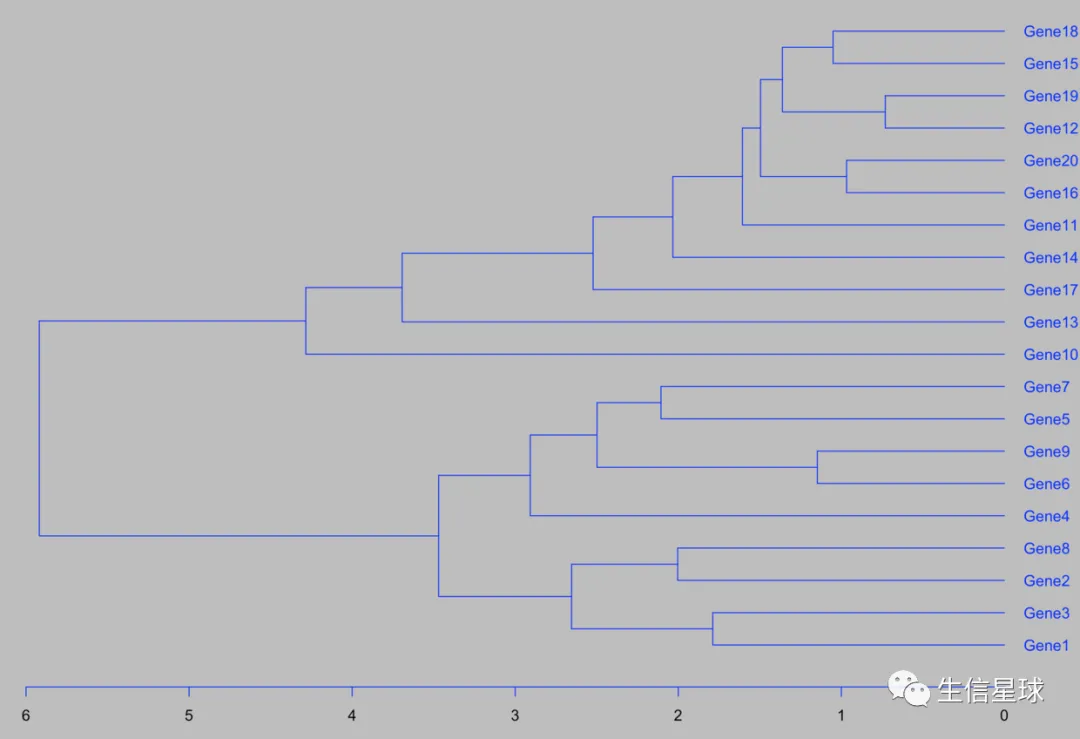
> rownames(diy1)[p1$tree_row[["order"]]]
[1] "Gene1" "Gene3" "Gene2" "Gene8" "Gene4" "Gene6" "Gene9" "Gene5" "Gene7" "Gene10" "Gene13" "Gene17"
[13] "Gene14" "Gene11" "Gene16" "Gene20" "Gene12" "Gene19" "Gene15" "Gene18"
自己聚类是这样的:
my_hclust_gene <- hclust(dist(diy1), method = "complete")
as.dendrogram(my_hclust_gene) %>%
plot(horiz = TRUE)
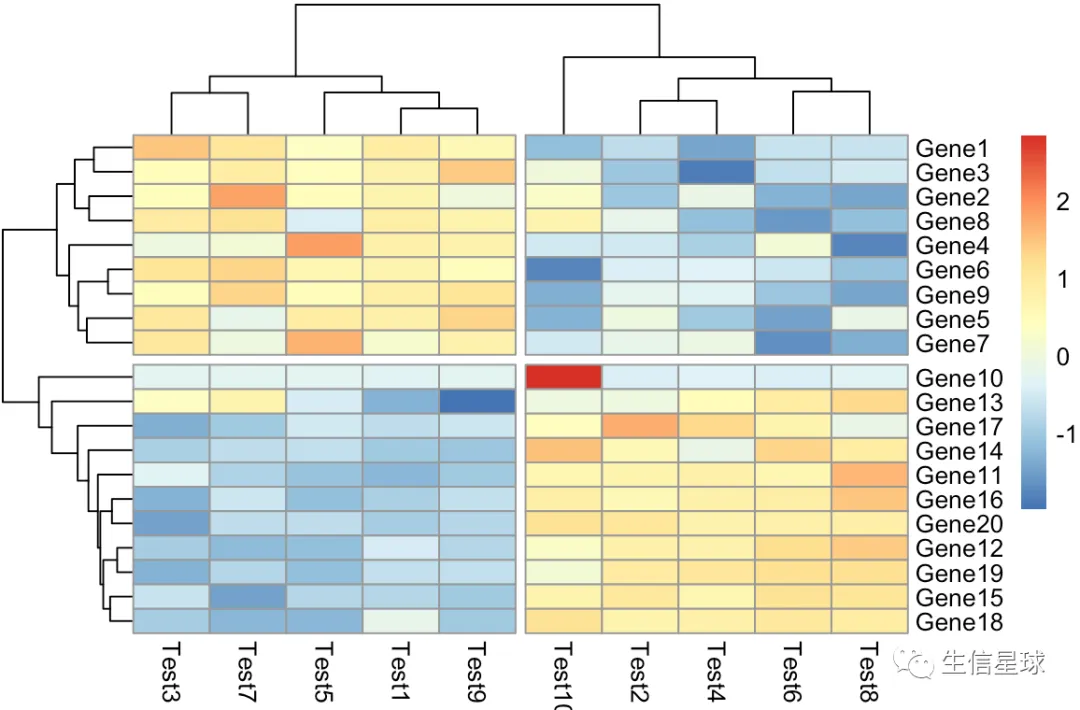
第五个图:把树切分
pheatmap(diy1,cutree_rows = 2,cutree_cols = 2)
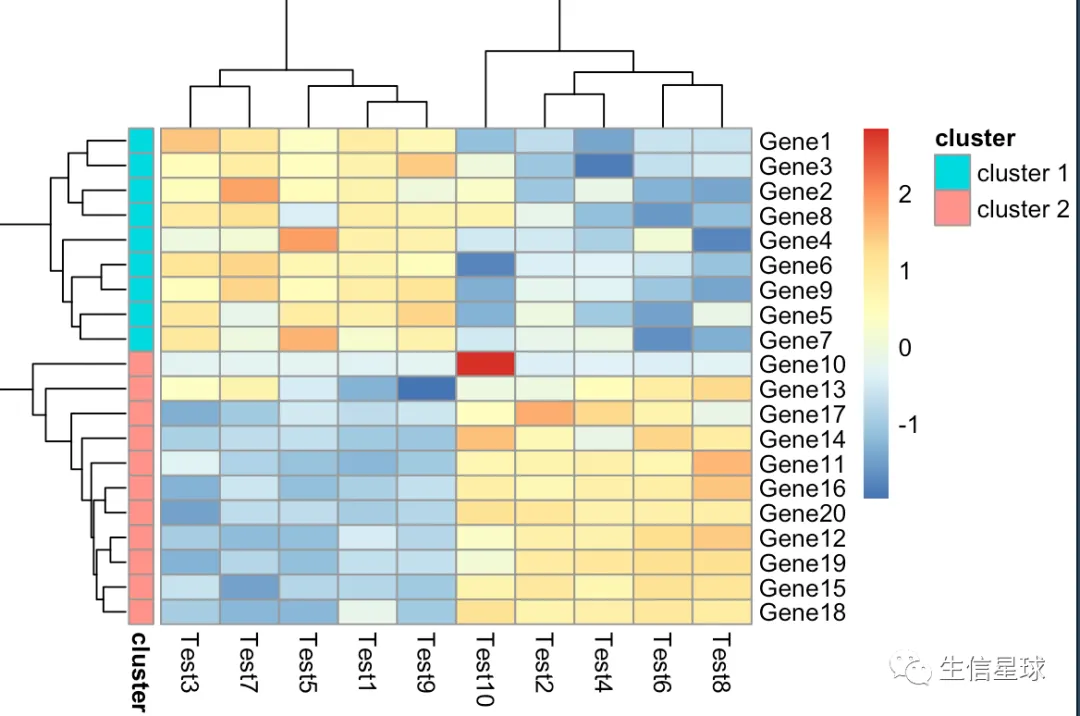
第六张图:自己给切分后的基因上色
# cutree_rows = 2就做了下面这行代码的事情
gene_anno <- cutree(tree = as.dendrogram(my_hclust_gene), k = 2)
gene_anno <- data.frame(cluster = ifelse(gene_anno == 1, "cluster1", "cluster2"))
> table(gene_anno)
my_gene_col
cluster 1 cluster 2
9 11
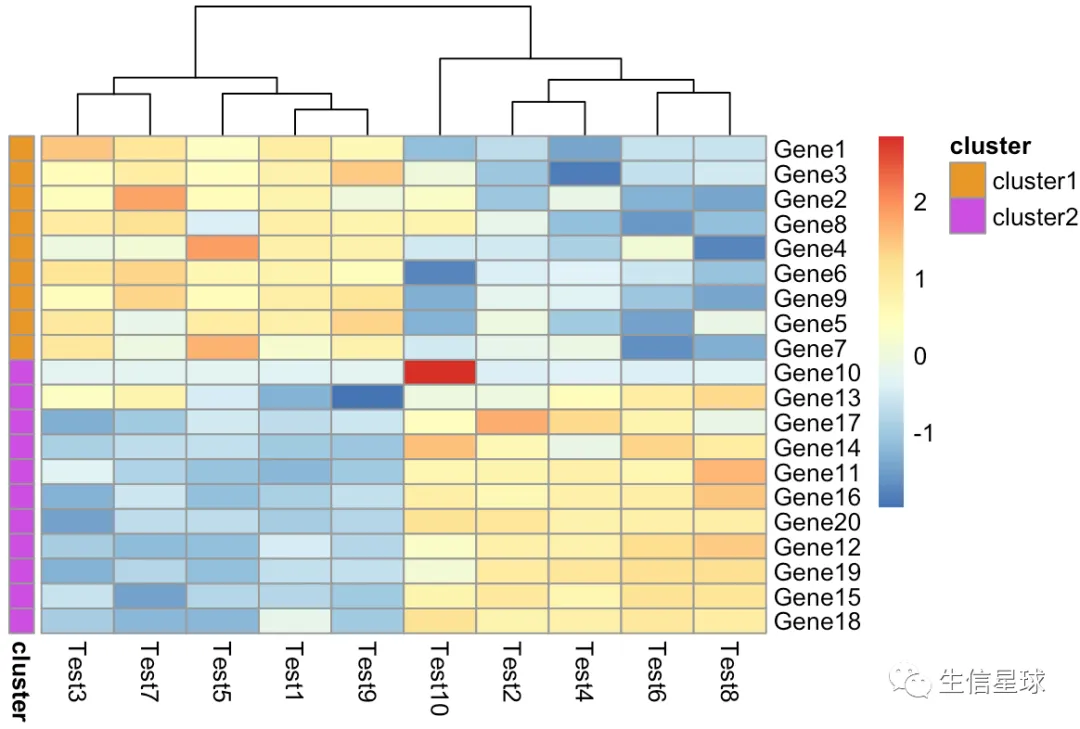
pheatmap(diy1, annotation_row = gene_anno)
修改颜色
需要设置一个list,如果有列的注释信息,也是添加到这个my_colour的list中
my_colour = list(
cluster = c(cluster1 = "#e89829", cluster2 = "#cc4ee0")
)
pheatmap(diy1,
annotation_colors = my_colour,
annotation_row = my_gene_col,
treeheight_row = 0)