185-KEGGREST包学习(附送KEGG复习)
刘小泽写于2020.5.10 这个包就是帮助我们理解KEGG数据库的,会了它就可以知道哪个基因属于哪个通路,以及不同通路中包含的基因
1 前言
在正式学习这个包之前,还是先补充一点KEGG数据库的知识吧
KEGG数据库是一个综合数据库,其中我们最常用的就是通路(PATHWAY)了,主要介绍了细胞生化过程(还有图注),如细胞代谢、细胞周期、膜转运、信号传递。但PATHWAY只能算上是一个小分支,全部的数据库可以看下面:
1.1 首先,KEGG主要有4大分类
系统信息 Systems information
基因组信息 Genomic information
化学信息 Chemical information
健康信息Health information
后来健康信息+药物标签 Drug labels = KEGG MEDICUS
然后每个分类下面又有自己的子数据库,比如常用的pathway数据库就是在系统信息之下的
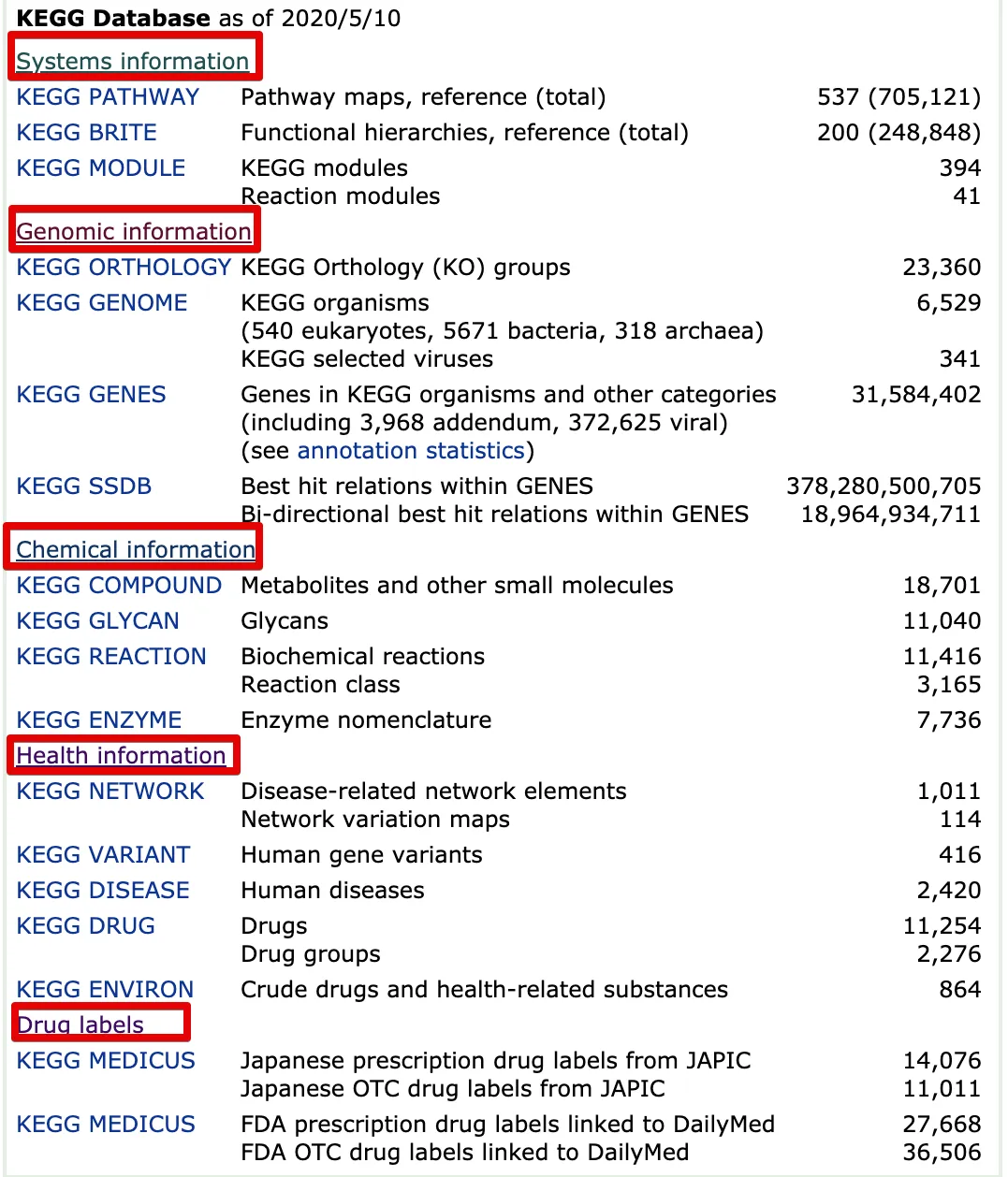
1.2 每个分类都有各自的颜色
这个图片有PDF版哦,而且其中例子的链接都是能点进去的,方便熟悉理解 公众号后台回复:KEGG 即可获得
Kanehisa 实验室 于1995年建立KEGG,子数据库的时间也在下面图片
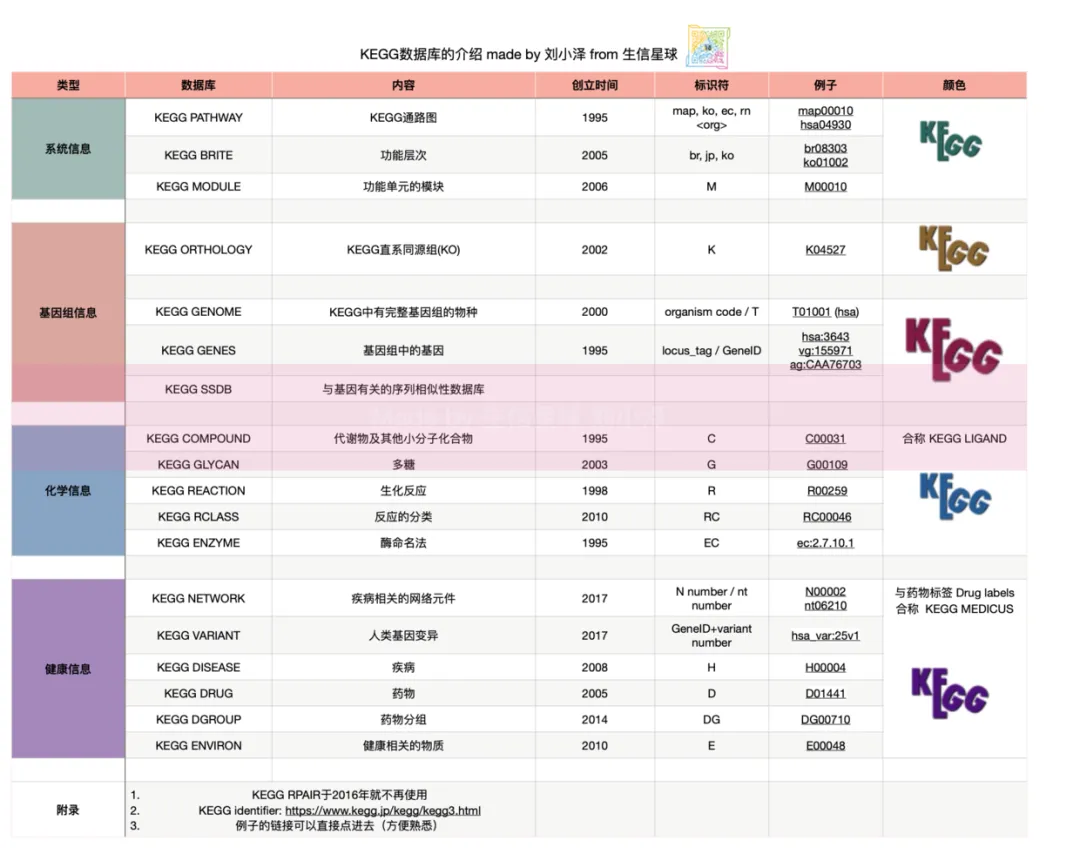
1.3 每个分支有各自的作用
来自官网的描述:https://www.kegg.jp/kegg/kegg1a.html
KEGG数据库是生物系统的数据重现,由基因、蛋白质(基因组信息)和化学物质(化学信息)的分子模块组建而成。这些分子模块之间构成了相互作用的关系网络(系统信息)。另外还包含了对生命活动有影响的疾病和药物资源(健康信息和药物信息)。
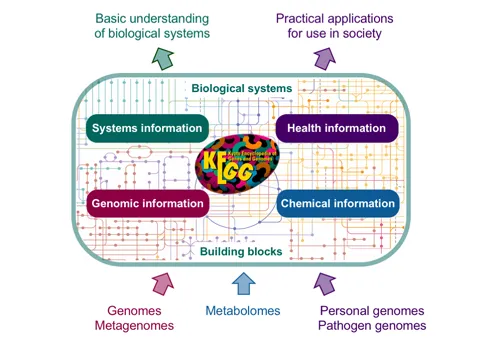
1.4 比较常用的子数据库
1.4.1 KEGG PATHWAY代谢通路数据库
来自:https://www.kegg.jp/kegg/kegg3a.html
包含了许多手工绘制或计算机模拟的代谢通路,包含了细胞过程、遗传信息、新陈代谢、疾病、药物开发等的分子互作和反应网络图。
每个pathway map都是通过2-4个字母的前缀 + 5位数字组成
其中前缀有5种类型:
- map - Reference pathway:手工绘制;根据已有的知识绘制的全面的供参考的代谢图,也是为后面几种的基础。图中的小框都是白色(后期可以填充);每个节点表示某个基因、该基因编码的酶或者酶参与的反应,可以点击其中的链接查看详细信息
- org - Organism-specific pathway map:计算机模拟;物种特有的代谢通路;绿色小框表示物种特有的基因或酶,点击绿色框的基因会看到更详细的信息
- ko - Reference pathway (KO):计算机模拟,节点只代表基因;点击蓝色框会看到直系同源基因信息
- ec - Reference pathway (EC):计算机模拟,节点只代表相关的酶
- rn - Reference pathway (Reaction):计算机模拟,节点只代表参与的某个反应、反应物及反应类型
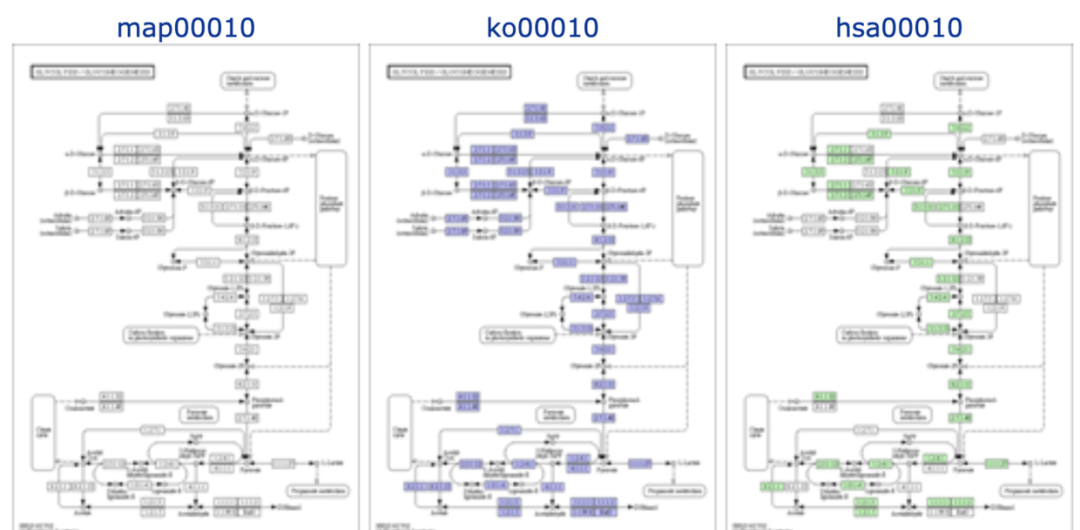
更详细的图片小物件解释可以看官网:https://www.genome.jp/kegg/document/help_pathway.html 【来自知乎白墨的翻译图】
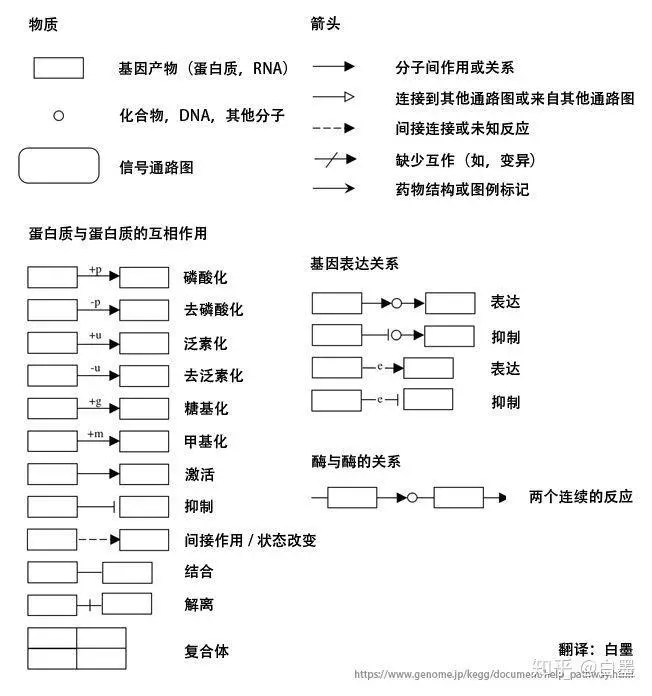
1.4.2 KEGG ORTHOLOGY 直系同源数据库
这个数据库是基于:同源基因功能相似。会把每个功能已知的基因和其他同源的基因归为一类,即一个KO号,它不分物种,比如ko00010(https://www.kegg.jp/kegg-bin/show_pathway?ko00010)就是糖酵解
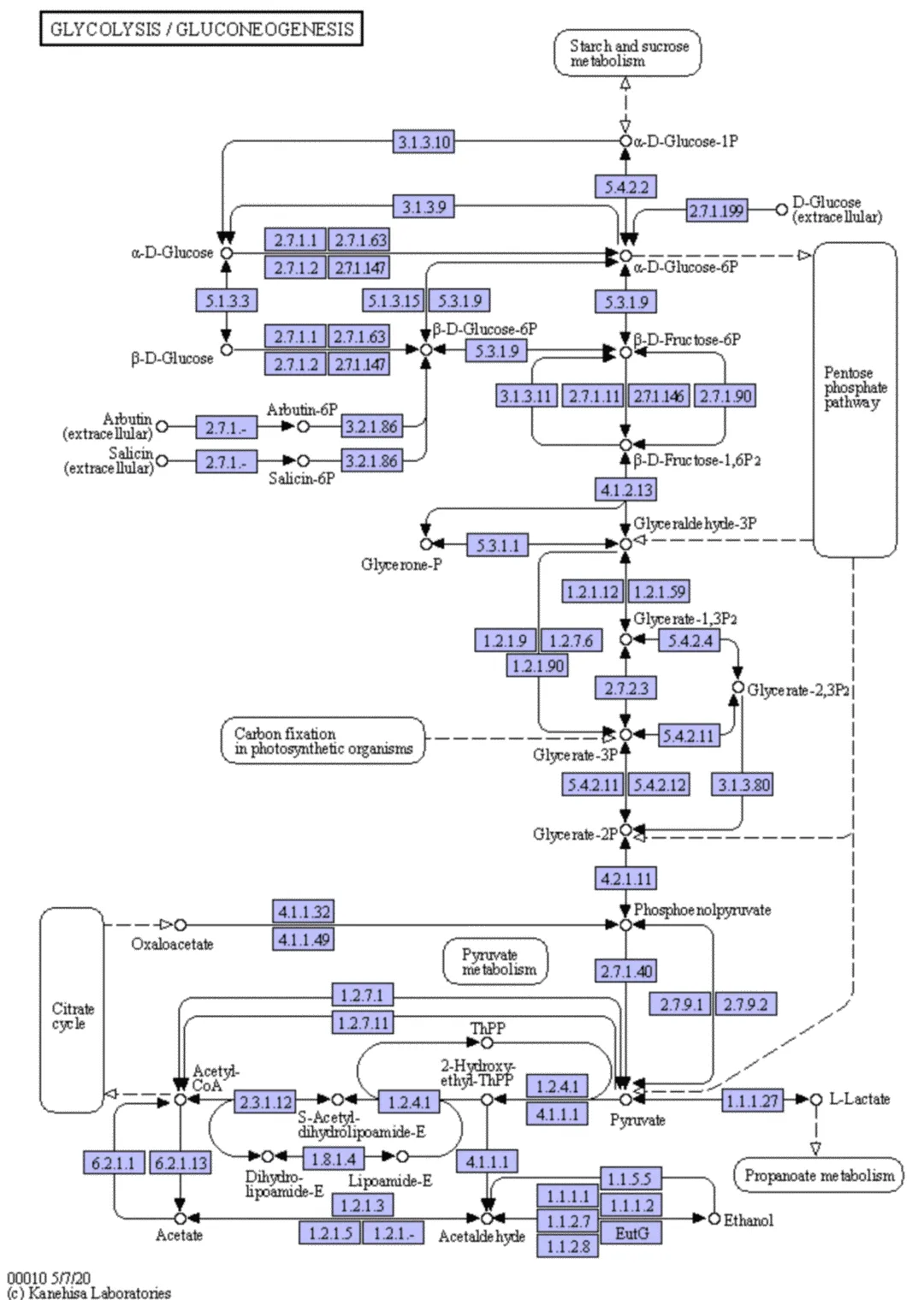
图中又这么几个形状:
圆角矩形:其他的KO通路
**方框:**如果把鼠标悬停在方框上,就会弹出K+数字。比如这里的K02777、K02779等,表示所有物种的一个同源基因

至于这个同源基因在各个物种中分别是什么,可以看详情,例如K02777(https://www.kegg.jp/dbget-bin/www_bget?ko:K02777)
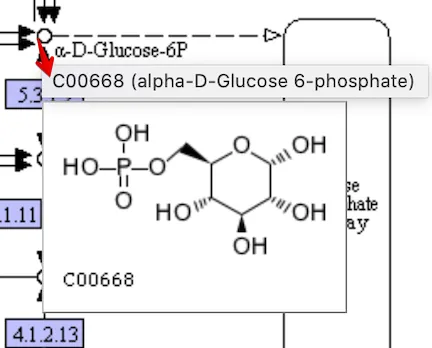
**小圆圈:**如果把鼠标悬停在方框上,会弹出化合物信息(C符号)
好,前言就这么多,开始学习这个R包啦 帮助文档在:https://bioconductor.org/packages/release/bioc/vignettes/KEGGREST/inst/doc/KEGGREST-vignette.html
2 正文开始
KEGGREST包可以获取
KEGG REST API ,之前获取KEGG数据可以用KEGGSOAP获取(但2012.12.31后就不再支持),现在KEGGREST是替补。不过相比之前的 KEGGSOAP ,使用更加简单功能也更多。
使用KEGG REST API 时的主要操作是: info, list, find, get, conv, and link
现在使用这个R包,主要操作是:keggInfo(), keggList(), keggFind(), keggGet(), keggConv, and keggLink()
2.1先看看能获取到数据库的什么内容
> library(KEGGREST)
> listDatabases()
[1] "pathway" "brite" "module"
[4] "ko" "genome" "vg"
[7] "ag" "compound" "glycan"
[10] "reaction" "rclass" "enzyme"
[13] "disease" "drug" "dgroup"
[16] "environ" "genes" "ligand"
[19] "kegg"
2.2 利用keggList()获取全部描述信息
2.2.1 获取全部物种信息
org <- keggList("organism")
head(org)
## T.number organism species
## [1,] "T01001" "hsa" "Homo sapiens (human)"
## [2,] "T01005" "ptr" "Pan troglodytes (chimpanzee)"
## [3,] "T02283" "pps" "Pan paniscus (bonobo)"
## [4,] "T02442" "ggo" "Gorilla gorilla gorilla (western lowland gorilla)"
## [5,] "T01416" "pon" "Pongo abelii (Sumatran orangutan)"
## [6,] "T03265" "nle" "Nomascus leucogenys (northern white-cheeked gibbon)"
## phylogeny
## [1,] "Eukaryotes;Animals;Vertebrates;Mammals"
## [2,] "Eukaryotes;Animals;Vertebrates;Mammals"
## [3,] "Eukaryotes;Animals;Vertebrates;Mammals"
## [4,] "Eukaryotes;Animals;Vertebrates;Mammals"
## [5,] "Eukaryotes;Animals;Vertebrates;Mammals"
## [6,] "Eukaryotes;Animals;Vertebrates;Mammals"
2.2.2 获取全部通路信息
如果直接用keggList("pathway") 得到的是map的通路
> head(keggList("pathway"))
path:map00010
"Glycolysis / Gluconeogenesis"
path:map00020
"Citrate cycle (TCA cycle)"
path:map00030
"Pentose phosphate pathway"
path:map00040
"Pentose and glucuronate interconversions"
path:map00051
"Fructose and mannose metabolism"
path:map00052
"Galactose metabolism"
# 总共有537条map通路
length(keggList("pathway"))
如果想获得某物种的全部通路(比如人)
res <- keggList("pathway", "hsa")
length(res)
# 337条人的通路
如果想根据一些关键词挑选通路【比较重要】
比如我现在想看人类的通路中和细胞凋亡、分化有关的
options(stringsAsFactors = F)
pat=keggList("pathway", "hsa")
pat=as.data.frame(res)
pat$id=stringr::str_split(rownames(pat),":",simplify = T)[,2]
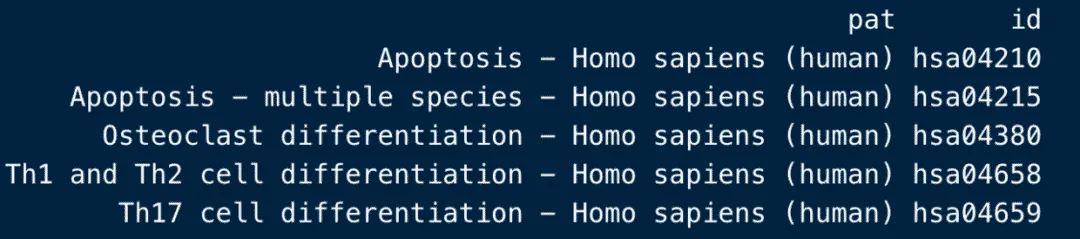
# 看到我们这里设置了不区分大小写(这里写apoptosis,但识别出来的是Apoptosis);并且不用写全单词(这里写的是different,但会识别出differentiation)
p=pat[grepl('different|apoptosis',pat[,1],ignore.case=T),]
2.3 利用keggGet()提取指定信息
来查看一个人类的基因和一个大肠杆菌的基因
query <- keggGet(c("hsa:10458", "ece:Z5100"))
length(query)
# 2
2.3.1 看看人类这个基因
names(query[[1]])
## [1] "ENTRY" "NAME" "DEFINITION" "ORTHOLOGY" "ORGANISM"
## [6] "PATHWAY" "BRITE" "POSITION" "MOTIF" "DBLINKS"
## [11] "STRUCTURE" "AASEQ" "NTSEQ"
2.3.2 看看这个基因涉及的通路
> query[[1]]$PATHWAY
hsa04520
"Adherens junction"
hsa04810
"Regulation of actin cytoskeleton"
hsa05130
"Pathogenic Escherichia coli infection"
hsa05135
"Yersinia infection"
和网页一致:https://www.kegg.jp/dbget-bin/www_bget?hsa:10458

2.3.3 获得蛋白序列
keggGet(c("hsa:10458", "ece:Z5100"), "aaseq")
## AAStringSet object of length 2:
## width seq names
## [1] 552 MSLSRSEEMHRLTENVYKTIMEQ...DLSAQGPEGREHGDGSARTLAGR hsa:10458 K05627 ...
## [2] 248 MLNGISNAASTLGRQLVGIASRV...SGLPPLAQALKDHLAAYEQSKKG ece:Z5100 K12786 ...
2.3.4 获得DNA序列
keggGet(c("hsa:10458", "ece:Z5100"), "ntseq")
## DNAStringSet object of length 2:
## width seq names
## [1] 1659 ATGTCTCTGTCTCGCTCAGAGGA...CCCGCACCCTGGCTGGAAGATGA hsa:10458 K05627 ...
## [2] 747 ATGCTTAATGGAATTAGTAACGC...ATGAGCAATCGAAGAAAGGGTAA ece:Z5100 K12786 ...
看到上面👆蛋白和DNA提取的结果都是:DNAStringSet对象,所以看看这个对象怎么用:https://uclouvain-cbio.github.io/WSBIM1322/sec-biostrings.html
如果想提取序列可以直接as.character(),例如
as.character(keggGet(c("hsa:10458", "ece:Z5100"), "ntseq")[1])
# 提取hsa:10458的序列

2.3.5 如果想看单个通路的图片
png <- keggGet("hsa05130", "image")
t <- tempfile()
library(png)
writePNG(png, t)
if (interactive()) browseURL(t)
2.3.6 根据通路找基因【常用】
先看单个通路的操作
# 首先得到全部的基因结果(其中会包含Entrez ID和Symbol以及详细的基因信息)
all=keggGet('hsa04210')[[1]]$GENE

entrez=all[seq(1,length(all),2)]
symbol=stringr::str_split(all[seq(2,length(all),2)],";",simplify = T)[,1]
id=data.frame(entrezID=entrez,symbolID=symbol)
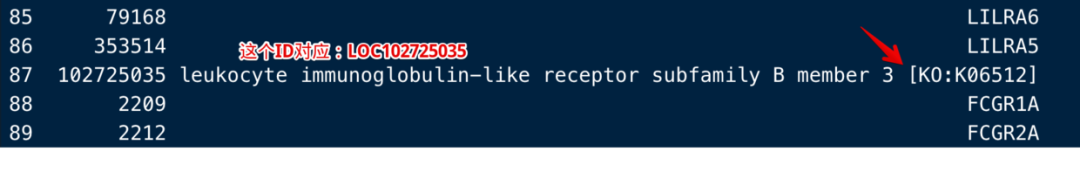
如果symbol这里有对应不上的,会给出Gene Description,例如:
然后看多个通路的循环操作【重要】
我们之前在2.2.2中根据一些关键词挑选了通路,并保持为p
p=pat[grepl('different|apoptosis',pat[,1],ignore.case=T),]
> length(p)
[1] 5
现在,我们就对p中这5个通路进行循环操作
# 之前p是字符串,现在变成列表,用于存储一会得到的各个结果
p=as.list(p)
pp=lapply(p, function(x){
# 其实还是像单个处理一样
all=keggGet(x)[[1]]$GENE
entrez=all[seq(1,length(all),2)]
symbol=stringr::str_split(all[seq(2,length(all),2)],";",simplify = T)[,1]
id=data.frame(entrezID=entrez,symbolID=symbol)
})
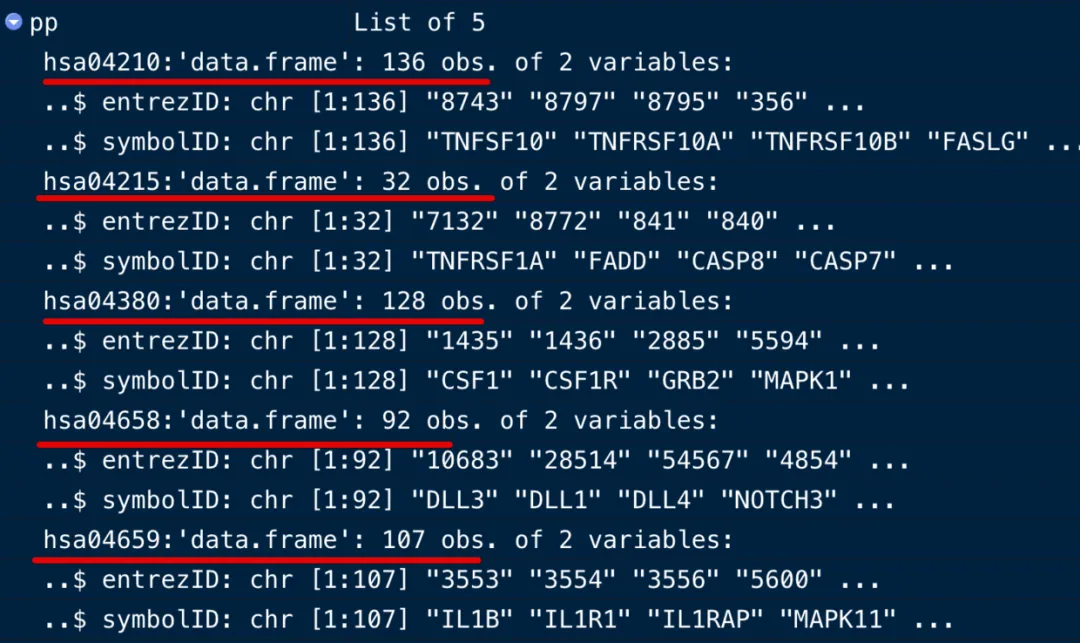
names(pp)=as.character(p)
最后会看到这5个通路中包含的基因数量
2.4 利用keggFind()搜索关键词
2.4.1 搜索基因信息中的两个词
比如ece:Z1464 这个基因详情中就包含"shiga”, “toxin"两个词
> head(keggFind("genes", c("shiga", "toxin")),3)
ece:Z1464
"stx2A; shiga-like toxin II A subunit encoded by bacteriophage BP-933W"
ece:Z1465
"stx2B; shiga-like toxin II B subunit encoded by bacteriophage BP-933W"
ece:Z3343
"stx1B; shiga-like toxin 1 subunit B encoded within prophage CP-933V"
#
搜索化学公式
head(keggFind("compound", "C7H10O5", "formula"))
## cpd:C00493 cpd:C04236 cpd:C16588 cpd:C17696 cpd:C18307 cpd:C18312
## "C7H10O5" "C7H10O5" "C7H10O5" "C7H10O5" "C7H10O5" "C7H10O5"
2.5 利用keggConv()转换KEGG ID
keggConv()前面参数是期待转换为的ID类型,后面参数是要转换的ID类型
例如将hsa:10458转为ncbi-proteinid
keggConv("ncbi-proteinid", c("hsa:10458", "ece:Z5100"))
## hsa:10458 ece:Z5100
## "ncbi-proteinid:NP_059345" "ncbi-proteinid:AAG58814"
2.6 利用keggLink()跨KEGG数据库搜索
2.6.1 比如找人类基因对应的所有的通路
head(keggLink("pathway", "hsa"))
## hsa:10327 hsa:124 hsa:125 hsa:126 hsa:127
## "path:hsa00010" "path:hsa00010" "path:hsa00010" "path:hsa00010" "path:hsa00010"
## hsa:128
## "path:hsa00010"
2.6.2 或者指定一个或多个基因(基因支持跨物种) 【比较常用】
keggLink("pathway", c("hsa:10458", "ece:Z5100"))
## hsa:10458 hsa:10458 hsa:10458 hsa:10458 ece:Z5100
## "path:hsa04520" "path:hsa04810" "path:hsa05130" "path:hsa05135" "path:ece05130"
3 重点总结
找全部通路
keggList("pathway")
根据基因找通路
keggLink("pathway", c("hsa:10458", "ece:Z5100"))
根据关键词找通路
options(stringsAsFactors = F)
pat=keggList("pathway", "hsa")
pat=as.data.frame(res)
pat$id=stringr::str_split(rownames(pat),":",simplify = T)[,2]
# 看到我们这里设置了不区分大小写(这里写apoptosis,但识别出来的是Apoptosis);并且不用写全单词(这里写的是different,但会识别出differentiation)
p=pat[grepl('different|apoptosis',pat[,1],ignore.case=T),]
根据通路找基因
# 这个p可以自己定义,也可以根据上面关键词找通路
p=pat[grepl('different|apoptosis',pat[,1],ignore.case=T),]
# 之前p是字符串,现在变成列表,用于存储一会得到的各个结果
p=as.list(p)
pp=lapply(p, function(x){
# 其实还是像单个处理一样
all=keggGet(x)[[1]]$GENE
entrez=all[seq(1,length(all),2)]
symbol=stringr::str_split(all[seq(2,length(all),2)],";",simplify = T)[,1]
id=data.frame(entrezID=entrez,symbolID=symbol)
})
names(pp)=as.character(p)